5. Vergleich und Auswahl von modernen HPLC-Säulen
Stefan Lamotte, BASF, Ludwigshafen, Stavros Kromidas, Saarbrücken, Frank Steiner ThermoScientific, München
Inhaltsverzeichnis
5.1 Trägermaterialien
5.1.1 Warum Kieselgel?
5.2 Stationäre Phasen für die HPLC- die geschichtliche Entwicklung
5.3 pH-Stabilität und Restriktionen beim Einsatz von Kieselgel
5.4 Die Kerneigenschaften von Umkehrphasen
5.4.1 Die Hydrophobie der Umkehrphasen
5.4.2 Die hydrophobe Selektivität
5.4.3 Die silanophile Aktivität
5.4.4 Die molekulare Formerkennung (shape selectivity)
5.4.5 Die polare Selektivität
5.4.6 Der Metallgehalt
5.5 Charakterisierung und Klassifizierung von Umkehrphasen
5.5.1 Über die Aussagekraft von Retentions- und Selektivitätsfaktoren bei Säulentests
5.5.2 Säulenvergleich, Vergleichskriterium: Ähnlichkeit von Selektivitäten
5.5.3 Zwei einfache Tests zur Charakterisierung von RP-Phasen
5.6 Vorgehensweise bei der Methodenentwicklung in der Praxis
5.6.1 Das Zusammenspiel zwischen mobiler und stationärer Phase
5.6.2 Welche Trennsäulen sollten verwendet werden und wie setze ich diese ein?
5.6.3 Was tun, wenn die Analyten sehr polar sind und auf den oben genannten Säulen keine Retention aufweisen?
5.6.3.1 AQ-Säulen, polare RP-Säulen und Ionenpaarchromatographie
5.6.3.2 Mixed Mode Säulen
5.6.3.3 Ionenausschlusssäulen/Ligandenaustauschchromatographie
5.6.3.4 HILIC (hydrophilic interaction liquid chromatography)
5.6.3.5 Poröser Kohlenstoff
5.7 Säulenscreening
5.8 Säulendatenbanken
5.1 Trägermaterialien
Aus der Historie heraus werden in der Flüssigchromatographie unterschiedlichste Materialien als stationäre Phasen eingesetzt. Dabei richtet sich deren Auswahl primär an drei Kerneigenschaften aus: chemische Stabilität, mechanische Stabilität und spezifische Oberfläche. Die chemische Stabilität ist wichtig, um eine möglichst große Kompatibilität mit den verwendeten mobilen Phasen zu erzielen. Damit steht und fällt auch die Möglichkeit die zu bestimmenden Analyte in Lösung zu bringen.
Der zweite ebenso wichtige Punkt ist die mechanische Stabilität. Da die HPLC in der Variante UHPLC unter Drücken bis über ca. 130-150 MPa (1300 -1500 bar, im Folgenden wird bar verwendet) stattfinden kann, sind die Materialien der stationären Phase u U hohen mechanischen Belastungen ausgesetzt. Dies hat zur Folge, dass nur bestimmte Trägermaterialien in Frage kommen.
Letztendlich ist es noch wichtig für die Flüssigchromatographie Basismaterialien mit einer hohen spezifischen Oberfläche einzusetzen. Dies ist notwendig, um ein gewisses Phasenverhältnis zwischen mobiler und stationärer Phase zu gewährleisten und somit eine entsprechende Beladbarkeit der stationären Phase sicher zu stellen. Eine solch hohe spezifische Oberfläche kann jedoch lediglich mit einem porösen Trägermaterial erreicht werden. Die Porosität hat wiederum einen Einfluss auf die mechanische Stabilität der stationären Phase.
Somit scheiden schon eine Vielzahl von Materialien aus und die Auswahl beschränkt sich auf Metalloxide wie SiO2, Al2O3, TiO2 und ZrO2, sowie quervernetzte Polymere auf Basis von Polystyrol/Divinylbenzol und Acrylaten, sowie Kohlenstoff.
Einen weiteren wichtigen Punkt in der Auswahl des Trägermaterials stellen dessen Herstellungspreis, sowie die Möglichkeit einer chemisch einfachen, flexiblen und kostengünstigen Oberflächenmodifizierung dar.
Aus diesen Gründen hat sich Kieselgel als Trägermaterial gegenüber allen anderen Materialien durchgesetzt und ist heute das mit Abstand am häufigsten verwendete Basismaterial für die HPLC. Dennoch gibt es einige Defizite bei der Verwendung von Kieselgel, so dass bei manchen Anwendungen durchaus andere Trägermaterialien zum Einsatz kommen. Dazu später mehr.
5.1.1 Warum Kieselgel?
Neben der mechanischen Stabilität, besitzt Kieselgel vor allem den Vorteil einer kostengünstigen und einfachen Synthese und einer einfachen, preiswerten und flexiblen Möglichkeit der Oberflächenmodifizierung. Ferner besteht bei Kieselgel im Gegensatz z. B. zu Polymermaterialien, die Möglichkeit die Porengröße bei der Synthese zielgerichtet und eng verteilt einzustellen. Dies ist ein wichtiger Punkt, um die Kinetik der Trennung zu optimieren, und somit schmale Elutionsbanden zu erzielen. Detaillierte Informationen zur Herstellung von Kieselgel, dessen Charakteristika und Chemie können im Buch von Isler [1] nachgelesen werden. Im Übrigen ist die Chromatographie nicht das Haupteinsatzgebiet von Kieselgel. Viel verbreiteter ist die Verwendung als Farbpigment, Füllstoff in der Kosmetik und Kunststoffindustrie, sowie als Adsorbens und als Träger für Katalysatoren (Stichwort: Zeolithe). Für die Praxis der HPLC ist es interessant, welche unterschiedlichen Ideen – man könnte fast schon sagen Philosophien - sich hinter der Charakteristik verschiedener Kieselgele verbergen, und welches der Zweck und Einsatzbereich dieser Kieselgele ist. Dazu ist es hilfreich sich die Wheeler Gleichung zu betrachten:

mit Vp: Porenvolumen in ml/g, SBET: Oberfläche in m2/g und D: Porengröße in Å
Daraus errechnen sich beispielsweise für ein typisches in der HPLC verwendetes Kieselgel mit einer Porengröße von 100 Å (10 nm) folgende Werte:

Und für eine typische für die HPLC verwendete Oberfläche von 300 m²/g folgende Möglichkeiten diese zu erhalten.

Was aber bedeutet das in der Praxis? Ein kleines Porenvolumen hat eine schnellere Diffusion zur Folge, was zu schmaleren Elutionsbanden (weniger gehinderte Diffusion) und damit zu höheren Bodenzahlen im direkten Vergleich zu größeren Porenvolumina führt. Allerdings dürfen die Porengrößen bei gleichzeitiger Verringerung des Porenvolumens nicht zu klein werden, damit es nicht zu Größenausschlusseffekten der Analyte kommt. Die chromatographisch aktive spezifische Oberfläche der stationären Phase ist immer der wichtigste Parameter des resultierenden Trägermaterials und steht daher bei der Entwicklung neuer Materialien immer im Zentrum der Optimierung.
Eine hohe spezifische Oberfläche der stationären Phase ist wünschenswert, da diese stärkere Wechselwirkungen der Analyte mit der stationären Phase mit sich bringt was häufig zu höherer Auflösung schwer zu trennender Analyte führt und auch Beladbarkeitsproblemen, die sich in asymmetrischen und breiten Elutionsbanden äußern, entgegenwirkt.
Außerdem sollte auch bedacht werden, dass eine Erhöhung der Porengröße auch eine verminderte mechanische Stabilität mit sich bringt, da das Gerüst des Trägermaterials dünner wird. Dies ist insbesondere bei Materialien für die UHPLC zu beachten, da dort Drücke bis 1500 bar erreicht werden können und die Säulen bei sogar noch höheren Drücken gepackt werden.
Je nach gewünschter Anwendung wurden daher unterschiedliche chromatographische Trägermaterialien entwickelt. Bei Kieselgel findet man Porenvolumina zwischen 0,3 und 1,5 ml/g, Porengrößen zwischen 60 Å und 300 Å und Oberflächen zwischen 80 m²/g und 500 m²/g. An späterer Stelle werden Beispiele genannt wo bei welchen Analyten welche Parameter eingesetzt werden.
5.2 Stationäre Phasen für die HPLC- die geschichtliche Entwicklung
Parallel zu Entwicklung des Kieselgels bezüglich Reinheit, Stabilität und Kinetik, muss auch die Entwicklung der Oberflächenmodifizierung betrachtet werden.
Am Anfang der HPLC wurden als Trägermaterialien irreguläre Kieselgele, die durch Polymerisation von Wasserglas hergestellt wurden, verwendet. Dies führte dazu, dass je nach Art der Einstellung des pH-Wertes der Lösungen und auch der Herkunft des verwendeten Wasserglases, der Metallgehalt und der pH-Wert der resultierenden Kieselgele stark unterschiedlich war. Die Acidität des Kieselgels hat wiederum entscheidenden Einfluss auf dessen Reaktivität, und somit bei modifizierten Kieselgelen auf deren Kerneigenschaften: Hydrophobie, hydrophobe Selektivität, silanophile Aktivität, molekulare Formerkennung und polare Selektivität.
Irreguläre Kieselgele wurden in den späten 1970er und frühen 1980er Jahren durch sphärische Kieselgele weitgehend abgelöst. Der Vorteil dieser lag primär in der höheren Packungsdichte und dem geringeren Gegendruck den diese Träger im Vergleich zu irregulären Materialien besitzen. Dies hat unmittelbar eine höhere mechanische Stabilität und damit eine höhere Lebensdauer der Trennsäule zur Folge. Dennoch hatten auch diese Kieselgele immer noch einen hohen, und auch schwankenden Metallgehalt, was zu Schwankungen in den Eigenschaften der resultierenden stationären Phasen führt. Stabile Retentionszeiten und gleiche Trenneigenschaften sind für den Einsatz der HPLC in der chemischen und pharmazeutischen Industrie jedoch unerlässlich.
Dennoch dauerte es noch etwa 10 Jahre bis Mitte 1980 erste hochreine Kieselgele auf der Basis der Polymerisation von Tetraethoxysilan (TES) eingeführt wurden. Der Metallgehalt (im wesentlichen Fe und Al) dieser Träger ist im ppm-Bereich. Damit ist es nun möglich, den Anforderungen der HPLC-Qualitätskontrolle nach zu kommen und sehr reproduzierbare Trennmedien zu produzieren. Diese so genannten Kieselgele der 3. Generation oder Typ B Kieselgele (wie sie im angelsächsischen Sprachraum gerne genannt werden) dominieren heute den Markt für Kieselgel.
Nichts desto trotz bleibt die Geschichte der Trägermaterialien für die HPLC nicht an diesem Punkt stehen und es gibt mehrere Weiterentwicklungen, die entweder auf eine kinetische Optimierung (s. Kap. 3 „Optimierung“) der Materialien oder auf eine Erweiterung des pH-Einsatzbereiches oder der Druckstabilität der Kieselgele abzielen. Bei der kinetischen Optimierung der Materialien werden im Wesentlichen die Diffusionswege für die mit der stationären Phase in Wechselwirkung tretenden Analyte verkürzt. Dies erfolgt primär durch Reduzierung der Partikeldurchmesser der Trägermaterialien. War es zu Beginn der 1990er Jahre durchaus noch üblich Träger mit einem Partikeldurchmesser von 5 µm zu verwenden, so war es Mitte der 1990er Jahre durchaus normal 3 µm Materialien gelegentlich einzusetzen. Mitte der 2000er Jahre – mit der Einführung der UHPLC - kamen dann breitbandig sub-2 µm Träger zum Einsatz. Ein wichtiger Trend in den 2010er Jahren zielt nun auf die Verwendung von Materialien mit unporösem Kieselgelkern, der von einer porösen Hülle ummantelt wird (im Nachfolgenden als solid core Material bezeichnet). Dabei kann durch die kürzeren Diffusionswege bei einem Gegendruck eines 3 µm Materials die Trenneffizienz eines 1,7 µm Materials erreicht werden. Dazu nutzen diese Materialien ein seit den 1970er Jahren entwickeltes, alt bekanntes Konzept. Schon die ersten Arbeiten mit HPLC nutzten bereits so genannte Porous Layer Beads [2, 3]. Allerdings wurden diese Materialien mit Partikelgrößen von etwa 50 µm damals als Verbesserung zu den üblichen irregulären Materialien (Fraktion 40 – 63 µm) verwendet. Der Nachteil lag in der deutlich geringeren Beladbarkeit (man hatte eine lediglich 1 µm dicke poröse Hülle um einen 50 µm dicken unporösen Glaskern, der den Vorteil der deutlich höheren Trenneffizienz nicht aufwog). Bei den neueren Ansätzen ist das Verhältnis Kern zu Hülle deutlich günstiger, so dass das Thema Beladbarkeit nur eine untergeordnete Rolle spielt (...). Die ersten solid core Materialien mit Partikelgrößen zwischen 2 µm und 3 µm zielten eindeutig darauf ab, eine verbesserte Alternative zu porösen sub 2 µm Materialien zu bieten. Der Anwender hat die Wahl entweder bei gleicher Trenneffizienz niedrigere Gegendrücke oder bei gleichem Gegendruck deutlich höhere Trenneffizienzen im Vergleich zu vollporösen sub 2 µm Materialien zu erreichen, indem er längere Trennsäulen einsetzen kann. Da die Elutionsbanden von solid core Materialien analog zu vollporösen sub-2 µm Materialien sehr schmal sind, ist es auch bei diesen Materialien - trotz anderslautender Aussagen auf Hochglanzbroschüren - nicht möglich, mit konventionellen HPLC-Anlagen zu arbeiten. Denn um die maximale Leistung dieser Trennsäulen zu nutzen, ist es zwingend notwendig, die Bandenverbreiterung außerhalb der Trennsäule zu optimieren und kleinvolumige Messzellen und höhere Datenaufnahmeraten zu verwenden. Aktuell wurden solid core Materialien mit größeren Partikelgrößen für den Einsatz in der HPLC entwickelt. Diese haben Partikelgrößen zwischen 4 µm und 5 µm, werden in klassische HPLC-Säulendimensionen gefüllt und weisen ebenfalls deutlich höhere Trenneffizienzen als vollporöse Materialien gleicher Partikelgröße auf. An dieser Stelle sollte jedoch erwähnt werden, dass Säulen, die mit solid core Materialien gefüllt sind, per se - wegen des unporösen Kerns - eine geringere totale Porosität besitzen, also eine höhere Packungsdichte aufweisen. In der Praxis bedeutet das, dass bei einem Vergleich einer vollporösen und einer solid core Säule gleicher Säulendimension, die bei gleichem Fluss (konkret: Volumenfluss) betrieben werden, die lineare Strömungsgeschwindigkeit in der solid core Säule höher ist, und somit ein um 20 bis 30% höherer Gegendruck im Vergleich zum entsprechenden voll porösen Material beobachtet wird. Will man die Trennsäulen bezüglich ihrer Eigenschaften wissenschaftlich korrekt vergleichen, muss bei vergleichbarer linearer Strömungsgeschwindigkeit gemessen werden, d.h. die vergleichbare solid core Säule muss bei geringeren Flussraten betrieben werden. Die lineare Strömungsgeschwindigkeit ist sehr einfach bestimmbar. Man dividiert die Säulenlänge durch die Durchfluss- oder Totzeit tM. Die Durchflusszeit kann aus dem Chromatogramm bestimmt werden. Es ist die Zeit bei der die Störung der Basislinie, die durch die Injektion der Probe verursacht, häufig auftritt. Die mit 4 µm bis 5 µm gepackten solid core Säulen können mit konventionellen HPLC - Apparaturen betrieben werden.
Neben diesen größeren solid core Partikeln sind auch kleinere sub-2 µm solid core Partikel mit Korngrößen von 1,7 µm und 1,3 µm kommerziell erhältlich. Deren durchaus vorhandenes theoretisches Potential bezüglich Trenneffizienz und Analysengeschwindigkeit kann jedoch in der Praxis nicht genutzt werden, da noch keine HPLC- Geräte zur Verfügung stehen, die die entsprechend notwendigen geringen Volumina (sowohl der Zuleitungen, als auch der Detektorzellen), sowie die erforderliche Datenaufnahmerate besitzen, um die auf diesen Säulen erzielten Trennungen verlustfrei detektieren und aufnehmen zu können. Die heutigen modernen UHPLC-Anlagen sind schon recht gut optimiert, jedoch reicht deren Leistung für diese Anwendungen bei weitem nicht aus. Hier sind die Gerätehersteller weiter gefordert. Wie bei allen Materialien, wird dringend empfohlen die Chargenreproduzierbarkeit gerade von solid core Phasen zu überprüfen.
Parallel zu der Entwicklung partikulärer Materialien, gibt es die Möglichkeit des Einsatzes monolithischer Trennsäulen. Diese seit Ende der 1990er Jahre sich entwickelnde Technik nutzt die Idee einer inversen Kieselgelstruktur mit dem gleichen Ziel: Beschleunigung der Kinetik. Der größte Vorteil dieser Technik ist der sehr geringe Gegendruck der Trennsäulen, die durchaus mit Säulen die mit sub-2 µm Trägern gefüllt sind vergleichbare Trenneffizienzen und schnelle Trennungen aufweisen [4]. Der größte Vorteil der monolithischen Trennsäulen liegt in der im Vergleich zu porösen Kieselgelsäulen höheren totalen Porosität des Materials. Dies bewirkt nicht nur den deutlich niedrigeren Gegendruck sondern auch, dass das Volumen der Trennsäule vergleichsweise höher ist. Das ist somit gerade umgekehrt wie bei den solid core Materialien. Für die Praxis bedeutet das, dass monolithische Trennsäulen eine deutlich stärkere Toleranz gegenüber Bandenverbreiterung außerhalb der Trennsäule zeigen, und somit auch auf konventionellen HPLC-Apparaturen ohne großen Optimierungsaufwand hohe Trenneffizienzen erreicht, und auch gemessen werden können. Ein weiterer Vorteil liegt in der besseren Verträglichkeit dieser Materialien gegenüber stark kontaminierten Proben. Diese Trennsäulen verschmutzen nicht so leicht wie vergleichbare partikuläre Materialien. Die Nachteile sollten jedoch auch nicht unerwähnt bleiben: Die Säulen sind auf einen Gegendruck von 200 bar limitiert, da die monolithischen Silicastäbe in einer PEEK-Ummantelung sitzen. Ferner gibt es auch nur wenige unterschiedlich selektive stationäre Phasen zu erwerben. Allerdings stellen monolithische Trennsäulen zur Zeit die robusteste und eleganteste Möglichkeit dar, schnelle Trennungen mit hinreichender Trenneffizienz (effektive 20.000 Böden auf einer Trennstrecke von 100 mm) zu erhalten.
5.3 pH-Stabilität und Restriktionen beim Einsatz von Kieselgel
Kieselgele sind der ideale Träger für stationäre Phasen in der HPLC, denn sie sind mechanisch stabil, lassen sich einfach und kostengünstig herstellen und sind chemisch relativ innert. Als Anhydride der Kieselsäure lösen sie sich jedoch im alkalischen Medium auf. Diesem Nachteil der Kieselgele wurde in der Geschichte der HPLC Rechnung getragen und mit verschiedenen Ansätzen entgegengewirkt. Die Entwicklung hochreiner Kieselgele war der erste Schritt. Besaßen die Träger der ersten Generation noch eine hohe Acidität, die von einem hohen Metallgehalt her rührte, so waren die Materialien der zweiten Generation deutlich reiner und dadurch pH-beständiger bei hohen pH-Werten. Weitere Ansätze eine höhere pH-Stabilität zu erzielen war das Beschichten der Kieselgele mit Metalloxiden wie Al2O3, TiO2 oder ZrO2, die die Auflösung des Kieselgels bei hohen pH-Werten verlangsamen [5]. Eine weitere Möglichkeit besteht darin, eine hohe Belegungsdichte bei den Umkehrphasen auf Kieselgelbasis zu erzielen. Bei hochbelegten Umkehrphasen wird die Auflösung des Kieselgels bei hohen pH-Werten erschwert, da die hydrophobe Oberfläche für die polaren Basen nur schwer zugänglich ist. Daher gilt: Je höher die Belegungsdichte des Trägermaterials und größer die verwendete Base, umso stabiler ist die stationäre Phase. Ein weiterer Ansatz eine höhere pH-Stabilität zu erhalten sind polymere Coatings um das Kieselgel, sowie die Einführung von Hybridmaterialien, also Komposite aus Tetraethoxysilan und verbrückten Organosilanen.
All diese Ansätze führen zwar zu einer leichten Verbesserung der pH-Stabilität um etwa eine pH-Werteinheit, jedoch wird dem Anwender fälschlicherweise suggeriert, er könne diese Säulen immer bei pH-Werten größer als 9 einsetzen. Dies ist so nicht richtig! Kieselgel ist auch als Hybridmaterial und mit Polymer ummantelt immer noch das Anhydrid der Kieselsäure und löst sich bei pH-Werten größer 7 auf. Durch eine hohe Belegungsdichte der stationären Phase, sowie durch ein Coating oder ein Organosilan-Hybrid kann diese Auflösung verlangsamt werden. Ferner wird durch die Verwendung von mobilen Phasen mit hohem Anteil an aprotische organischen Lösemitteln (z. B. Acetonitril) die Dissoziation der Basen und der Silanolgruppen des Kieselgels zurückgedrängt, was in der Praxis zu deutlich niedrigeren pH-Werten führt. (Vorsicht: Bis zu einem pH-Wert von ca. 8 steigt dieser nach der Zugabe von Methanol/Acetonitril, [6]). Findet man also in einer Hochglanzbroschüre eines Säulenherstellers die Stabilitätsstudie einer Kieselgel basierenden HPLC-Säule, die bei einem pH-Wert von 12 ohne Effizienzverlust 10.000 Säulenvolumina erreicht, so ist das nur möglich, wenn der organische Lösemittelanteil mindestens 70% beträgt, die Base sehr sperrig ist (z. B. Triethylamin oder Pyrrolidin) und bei Raumtemperatur gearbeitet wurde. Würde die gleiche Säule z. B. bei 60°C mit einer kleinen Base (Ammoniak oder Hydroxid) in mobilen Phasen die weniger als 10% organisches Lösemittel enthalten betrieben, wäre die Standzeit unter 500 Säulenvolumina. Daher ist hier Vorsicht geboten!
Muss dennoch in der Routine bei pH-Werten höher als 8 chromatographiert werden, so ist es evtl. sinnvoller, alternative Materialien auf Polymerbasis oder aus anderen Metalloxiden wie TiO2 oder ZrO2 oder aus reinem Kohlenstoff zu verwenden.
Es besteht jedoch auch noch die Möglichkeit eine so genannte „Opfersäule“ (oder Sättigungs- oder Konditionierungssäule) zu verwenden. Dies ist eine in der Praxis bewährte Maßnahme, die Standzeiten der Kieselgel basierenden Trennsäulen bei hohen pH-Werten der mobilen Phase zu erhöhen, ohne einen anderen Träger einzusetzen, was etwaige Selektivitätsprobleme mit sich bringen würde. Bei einer Opfersäule handelt es sich um eine kurze Säule, z. B. eine Vorsäule, die mit einem grobkörnigen Kieselgel gefüllt ist. Deren Partikel werden von der basischen mobilen Phase langsam aufgelöst und die mobile Phase sättigt sich mit dem aufgelösten Kieselgel, so dass die Trennsäule verschont wird. Es ist allerdings auch möglich, als Opfersäule einfach nur eine mit grobkörnigem Kieselgel gefüllte Säule zu verwenden. Die Opfersäule wird vor die Injektionseinheit (also vor Mischkammer und Probengeber) installiert, damit sie nicht das „Dwell Volume“ der Apparatur verändert. Warum ist es eigentlich überhaupt sinnvoll, bei hohen pH-Werten zu chromatographieren? Die Anwort ist einfach: Bei basischen pH-Werten liegen basische Analyte unprotoniert vor. Dadurch sind die hydrophoben Wechselwirkungen zwischen stationärer Phase und Analyt stärker. Die ionischen Wechselwirkungen werden schwächer und somit die Elutionsbanden der basischen Analyte schmäler und symmetrischer als würde man diese bei sauren oder neutralen pH-Werten chromatographieren. Ferner werden die basischen Analyte, da sie bei hohen pH-Werten nicht mehr ionisch sind, hydrophober und damit stärker auf Umkehrphasen retardiert, was häufig sehr günstig ist, weil damit eine Verschiebung der basischen Komponenten in ein anderes Elutionsfenster möglich ist. Außerdem zeigen Basen im ungeladenen Zustand eine deutlich höhere Beladbarkeit auf Umkehrphasen. [7].
Häufig sind die basischen Verbindungen im ungeladenen Zustand jedoch deutlich weniger stabil als im geladenen Zustand. Das ist eine bekannte Beobachtung, die bei Stabilitätsuntersuchungen in vielen Fällen festgestellt wird. In der Chromatographie werden manchmal Artefakte beobachtet, die auf die Zersetzung der Produkte hindeuten. Diese Tatsache wird oft verschwiegen. Daher ist bei Arbeiten bei hohen pH-Werten Vorsicht geboten. Dennoch ist die Variation des pH-Wertes, neben der Auswahl der stationären Phase, der wichtigste Optimierungsparameter bei der Trennung polarer bzw. ionischer/ionisierbarer Komponenten.
5.4 Die Kerneigenschaften von Umkehrphasen
Die Eigenschaften, die die Selektivität der Umkehrphasen im Wesentlichen beeinflussen, sind Hydrophobie, hydrophobe Selektivität, silanophile Aktivität, molekulare Formerkennung (shape selectivity), polare Selektivität und Metallgehalt. Diese Eigenschaften sind für alle verschiedenen Trägermaterialien unterschiedlich und bestimmen daher wesentlich die Trennung.
5.4.1 Die Hydrophobie der Umkehrphasen
Die Hydrophobie der Umkehrphase gibt an, wie stark ein primär via hydrophober Wechselwirkungen mit der stationären Phase interagierender Analyt retardiert wird. Kurz: Je stärker die Retentionskraft der stationären Phase, umso höher die Hydrophobie. Die Hydrophobie einer Trennsäule wird durch die Oberfläche des Trägermaterials, dessen Kohlenstoffgehalt und der Belegungsdichte des Trägermaterials bestimmt. Phasen die gleiche Hydrophobie haben, können jedoch durchaus recht unterschiedliche Trennungen liefern (selbst für homologe Reihen)! Daher sollte auch die hydrophobe Selektivität berücksichtigt werden, die für die Trennung bedeutender als die Hydrophobie selbst ist [8].
5.4.2 Die hydrophobe Selektivität
Die hydrophobe Selektivität ist die Methylengruppenselektivität der stationären Phase, d.h. die Selektivität zweier überwiegend auf Grund ihrer Hydrophobie retardierter Verbindungen, die sich durch eine Methylengruppe unterscheiden. Diese Selektivität ist z. B. im Falle von neutralen, hydrophoben Analyten zum Vergleich von Säulen und der Beurteilung ihrer Gleichheit heranzuziehen und besitzt eine deutlich höhere Aussagekraft als die in 5.1 behandelte Hydrophobie, s. auch weiter unten
5.4.3 Die silanophile Aktivität
Kieselgel besitzt ca. 9 µmol/m² Silanolgruppen auf der Oberfläche [1]. Setzt man Kieselgel mit C18-Silan um, gelingt es, ca. 3 µmol/m² dieser Silanolgruppen zur Reaktion zu bringen. Verbleiben noch ca. 6 µmol/m². Weitere Reaktionen mit kürzerkettigen Silanen (auch „Endcapping“ genannt) - meist Trimethylsilyl funktionalisierte Silane - führt zu einer weiteren Umsetzung von ca. 3 µmol/m² Silanolgruppen, so dass bei der fertigen Umkehrphase noch ca. 3 µmol/m² Silanolgruppen verbleiben, die nicht weiter umsetzbar sind, die aber sehr wohl Wechselwirkungen mit Silanolgruppen-affinen Analyten eingehen können, die zur Selektivität der entsprechenden Umkehrphase beitragen. Man spricht von der so genannten „silanophilen Aktivität“ der Umkehrphasen. Die Silanolgruppen-affinen Verbindungen sind meist Basen, die mit den sauren Silanolgruppen über ionische Wechselwirkungen interagieren. Diese Wechselwirkungen sind energetisch deutlich stärker als die üblichen hydrophoben Wechselwirkungen von Umkehrphasen, was in der Regel zu breiteren Elutionsbanden von basischen Analyten auf klassischen Umkehrphasen führt. Daher richtet sich das Bestreben der Hersteller bei der Entwicklung neuer Umkehrphasen primär auf eine Minimierung der silanophilen Aktivität. Diese ist auch der kritischste Parameter bei der reproduzierbaren Herstellung von Umkehrphasen. Die silanophile Aktivität einer Umkehrphase wird durch folgende Parameter bestimmt: Herstellungsprozess des Kieselgels, Belegungsdichte der stationären Phase, Art des verwendeten Silanes (reaktive und funktionelle Gruppen des Silanes). An dieser Stelle sei jedoch angemerkt, dass es nicht in allen Fällen Ziel führend ist, stationäre Phasen mit niedriger silanophiler Aktivität zu verwenden. Sind beispielsweise keine basischen Verbindungen in der Probe vorhanden, können Silanolgruppen durchaus positiv zur Selektivität einer stationären Phase beitragen. So werden häufig Wasserstoffbrückenbindungen der Silanolgruppen zu Carbonylgruppen beobachtet ohne die manche Trennungen gar nicht möglich wären.
5.4.4 Die molekulare Formerkennung (shape selectivity)
Umkehrphasen können auch zwischen räumlichen Anordnungen von Analyten in Lösung unterscheiden, also ob Analyten planar oder gewinkelt vorliegen, was z. B. häufig bei cis-, trans-Isomeren der Fall ist. Diese Eigenschaft nennt man molekulare Formerkennung („shape selectivity“). Für die Stärke der molekularen Formerkennung der Umkehrphase sind im Wesentlichen drei Parameter der stationären Phase verantwortlich: Belegungsdichte der Umkehrphase, Kettenlänge der Umkehrphase, zusätzliche polare Wechselwirkungen und Porengröße des Kieselgels. Es gilt:
- je höher die Belegungsdichte, umso höher die molekulare Formerkennung
- je länger die Alkylkette, umso höher die molekulare Formerkennung
- je weiter die Poren, umso höher die molekulare Formerkennung
- je mehr zusätzliche polare Wechselwirkungen, umso höher die molekulare Formerkennung
Warum ist das so?
Die Antwort liegt auf der Hand. Die molekulare Formerkennung einer stationären Phase steht und fällt mit der stärkeren Möglichkeit des planareren Analyten mit der stationären Phase in Wechselwirkung zu treten und der gleichzeitigen Diskriminierung der Wechselwirkung für den in gewinkelter Form vorliegenden Analyten. Ist die Belegungsdichte hoch, so kann nur das planarere Molekül, mit all seinen Wechselwirkungsstellen komplett in die Poren der stationären Phase eindringen und erfährt somit eine stärkere Retention als das gewinkelte Molekül. Analog ist die Sachlage bei längeren Alkylketten der stationären Phase. Hier ziehen sich die Alkylketten gegenseitig stärker an, je flexibler (länger) sie sind, umso engere „Spalten“ bilden sie, in die nur die planaren Moleküle eindringen können. Die Abhängigkeit von der Porengröße resultiert aus der Tatsache, dass über 99% der chromatographischen Oberfläche sich innerhalb der Kieselgelpartikel befindet. Sind die Poren zu klein, ist es schlichtweg unmöglich, die erforderliche Belegungsdichte für eine hinreichende molekulare Formerkennung zu erreichen. Daher haben die meisten „shape“-selektiven Umkehrphasen Porengrößen die größer als 150 Å (15 nm) sind. Auch hier bestätigen selbstverständlich Ausnahmen die Regel.
5.4.5 Die polare Selektivität
Unter polarer Selektivität versteht man die Retentionsunterschiede polarer Analyte relativ zu neutralen Analyten im Vergleich zu klassischen Umkehrphasen. Auf polar selektiven stationären Phasen werden im Vergleich zu klassischen Umkehrphasen Säuren stärker und Basen leicht schwächer retardiert. Die polare Selektivität wird durch polare Gruppen (Amid, Carbamat, Harnstoff, Diol, usw.) bewirkt, die auf die Oberfläche des Kieselgels aufgebracht werden, und die zusätzliche Wechselwirkungen mit den polaren Analyten eingehen können. Polar selektive Umkehrphasen sind die erste Alternative falls klassische Umkehrphasen nicht funktionieren, aber dazu später mehr.
5.4.6 Der Metallgehalt
Der Metallgehalt wird durch die Herstellung des Kieselgels bestimmt. Je nach verwendetem Ausgangsmaterial und Gerätschaften ist er höher oder niedriger. Die ersten Generationen Kieselgel wurden aus Wasserglas hergestellt. In diesen Kieselgelen war der Gehalt an Metallen relativ hoch. Vor allem Aluminium und Eisen sind darin zu finden. Was ist nun aber so schlimm daran, wenn sich Metallverunreinigungen im Kieselgel befinden? Eigentlich sollte das lediglich Probleme mit einigen wenigen Analyten, die Wechselwirkungen mit diesen Metallen eingehen können (z. B. Komplexbildner oder einige Proteine) machen. Diese sollten zusätzliche Wechselwirkungen mit der stationären Phase eingehen, was in aller Konsequenz breite unsymmetrische Elutionsbanden zur Folge hat. Jedoch beeinflussen die Metalle auch die Acidität des Kieselgels und damit die silanophile Aktivität (siehe 5.2) und von daher auch die Reaktivität der Silanolgruppen mit den Silanen bei der Oberflächenmodifizierung. Auch das wäre nicht so schlimm, wäre der Metallgehalt der Ausgangskieselgele immer gleich. Dies ist jedoch nicht der Fall. Somit ist die Batch zu Batch Reproduzierbarkeit von stationären Phasen auf der Basis von nicht metallfreien Kieselgelen (die angelsächsische Literatur spricht häufig von Typ A Kieselgelen) sehr viel schlechter, als die von metallfreien Kieselgelen (so genannten Typ B Kieselgelen). Aber auch hier gilt: Manche Trennungen bedürfen zusätzlicher Metallwechselwirkungen und es ist in vereinzelten Fällen durchaus möglich, dass die Trennung nur auf Typ A Kieselgelen funktioniert. So „schwört“ mancher Anwender heute immer noch auf „sein“ LiChrospher, Select B oder Spherisorb ODS 1...
5.5 Charakterisierung und Klassifizierung von Umkehrphasen
Aufgrund der oben genannten sechs Kerneigenschaften von Umkehrphasen können die stationären Phasen charakterisiert werden. Zu dieser Thematik gibt es eine große Vielzahl an Literatur [9-15]. Selbstverständlich kann man die synthetisierten stationären Phasen einer vollen physikochemischen Prüfung unterziehen (Stickstoffadsorptionsmessungen zur Bestimmung der spezifischen Oberfläche, des Porenvolumens und der Porengröße, CHN Analysen zur Bestimmung der Belegungsdichte der stationären Phase, Partikelgrößenmessungen, usw.). Jedoch sind all diese Charakterisierungen nicht Ziel führend, da am Ende lediglich die chromatographische Trennung zählt. Somit haben sich zur Charakterisierung und Klassifizierung von Umkehrphasen chromatographische Tests durchgesetzt, von denen hier einige repräsentativ ohne den Anspruch auf Vollständigkeit vorgestellt werden sollen.

Abbildung 1: Test polare Selektivität: neutrale (schwarz), saure (rot) und basische (grün) Analyte
Test | Polare Selektivität | |
Mobile Phase | ACN/50 mM Phosphatpuffer pH=3, 65/35 (v/v) | |
Temperatur | 40°C | |
Fluss | 1.0 ml/min (bei ID 4.0 mm Säulen) | |
Detektion | UV @ 254nm | |
Analyte | Analyt | Eigenschaft |
Uracil | Durchflusszeitmarker | |
Pyridin | Silanophile Aktivität | |
Phenol | Polare Selektivität | |
4-Butylbenzoesäure | Polare Selektivität | |
N,N-Dimethylanilin | Silanophile Aktivität | |
Toluol | Hydrophobie | |

Abbildung 2: Engelhardt Test: Vergleich klassiche Umkehrphase und Umkehrphase mit eingebundener polarer Gruppe
Test | Engelhardt Test | |
Mobile Phase | MeOH/Wasser 49/51 (w/w) bzw. (55/45 (v/v)) | |
Temperatur | 40°C | |
Fluss | 1.0 ml/min (bei ID 4.0 mm Säulen) | |
Detektion | UV @ 254nm | |
Analyte | Analyt | Eigenschaft |
Uracil | Durchflusszeitmarker | |
Anilin | Silanophile Aktivität | |
4-Ethylanilin | Silanophile Aktivität | |
N,N-Dimethylanilin | Silanophile Aktivität | |
Benzoesäureethylester | Kettenlängenselektivität | |
Toluol | Hydrophobie/hydrophobe Selektivität | |
Ethylbenzol | Hydrophobie/hydrophobe Selektivität | |

Abbildung 3: Neue Test; Vergleich Umkehrphase mit eingebundener polarer Gruppe und Fluorphase
Test | Neue Test | |
Mobile Phase | MeOH/20 mM Phosphatpuffer pH=7, 59,4/40,6 (w/w) (65/35 (v/v)) | |
Temperatur | 23°C | |
Fluss | 1.0 ml/min (bei ID 4.0 mm Säulen) | |
Detektion | UV @ 210nm | |
Analyte | Analyt | Eigenschaft |
Uracil | Durchflusszeitmarker | |
Propranolol | Silanophile Aktivität | |
Butylparaben | Polare Selektivität | |
Dipropylphthalat | Polare Selektivität | |
Naphthalen | Hydrophobie | |
Amitryptilin | Silanophile Aktivität | |
Acenaphthen | Hydrophobie | |
Es bleibt noch zu erwähnen, dass sich die Charakteristik der Trennsäulen bei deren Verwendung je nach Puffer, Additiv aber auch Probenmatrix verändert. Das hat für die Praxis mehrere Konsequenzen:
- Während einer Methodenentwicklung und –optimierung „sehen“ HPLC-Trennsäulen einige mobile Phasen verschiedenster pH-Werte und Puffer, sowie Additive. Wenn Sie am Ende einer Methodenoptimierung angekommen sind, charakterisieren Sie am Besten die Trennsäule mittels eines chromatographischen Tests. Dies erlaubt Ihnen etwaige spätere Abweichungen zu erkennen und rasch Ersatzlösungen zu finden. Zu dieser Thematik werden wir an späterer Stelle beim Thema Säulendatenbanken detaillierter eingehen.
- Während einer HPLC-Methodenvalidierung ist es zwingend notwendig, die fertig optimierte Methode an mehreren neuen Trennsäulen anderer Produktionschargen (i. d. R. testet man drei Säulen verschiedener Chargen) zu testen, ob die Trennung noch funktioniert. Ist dies der Fall können Sie guter Hoffnung sein, dass auch spätere Chargen funktionieren werden. Manchmal bedarf es dabei jedoch, dass die Trennsäulen mittels eines definierten Einspülprogramms vorkonditioniert werden müssen, bevor sie den Bedürfnüssen der Trennung Rechnung trägt. Daher ist die Überprüfung der Selektivität so wichtig.
5.5.1 Über die Aussagekraft von Retentions- und Selektivitätsfaktoren bei Säulentests
Vorbemerkung
Für die Retention der Analyte ist die Hydrophobie einer stationären Phase verantwortlich. Sie wird im Wesentlichen von der spezifischen Oberfläche und dem Kohlenstoffgehalt bestimmt, s. weiter oben. Was die Selektivität jedoch betrifft, spielen hydrophobe Wechselwirkungen in der RP-Chromatographie in der Regel eine weitaus geringere Rolle als allgemein angenommen wird. Bezüglich Selektivität sind ionische/polare Wechselwirkungen und sterische Aspekte – sofern sie zur Geltung kommen – dominantere Faktoren. Erst durch diese Einflussfaktoren können Analyte differenziert werden. Das ist auch der Grund warum eine Korrelation zwischen Retentions- und Selektivitätsfaktoren eher selten beobachtet wird, siehe weiter unten. Eine solche kann allenfalls bei einheitlichem RP-Mechanismus erwartet werden bei dem Van der Waals-Wechelwirkungen vorherrschen, was jedoch eher selten vorkommt (homologe Reihen oder einfache Analytpaare wie z. B. Toluol/Ethylbenzol), denn: die zu analysierenden Moleküle verfügen meistens über Gruppierungen, die zu ionischen/polaren Wechselwirkungen befähigt sind. Sogar bei hydrophoben Aromaten haben wir mit induzierten Dipolen zu tun, durch Mesomerie kann sich des Weiteren die Polarität erhöhen.
Auch unterscheiden sich sonst „ähnliche“ Moleküle in ihrer räumlichen Ausdehnung, deren sterische Eigenschaften beeinflussen ihr Diffusionsverhalten in den Poren der Phasenoberfläche. Als Beispiel kann man die Polarisierbarkeit von Molekülen aufgrund von Doppelbindungen oder die Starrheit bzw. Flexibilität in Abhängigkeit von der Lage der Substituenten am aromatischen Ring nennen.
Halten wir fest: In einem RP-System sind im Wesentlichen hydrophobe Wechselwirkungen für die Retention zuständig – bei ionischen Analyten naturgemäß auch Ionenaustausch, sofern ein solcher möglich ist. Für die Selektivität dagegen sind polare/ionische Wechselwirkungen entscheidend. Das sind in erster Linie Ionenaustausch sowie Wasserstoffbrückenbindungen und Dipol-Dipol Wechselwirkungen, ggf. auch Komplexbildung/Chelatisierung.
Kriterien zum Vergleich von Säulen
In der Literatur wird zum Vergleich von stationären Phasen als Kriterium vielfach die Hydrophobie („Hydrophobicity“) herangezogen. Dazu werden lnk-Werte (k=Retentionsfaktor) von typischen RP-Analyten an verschiedenen stationären Phasen verglichen: an „ähnlichen“ hydrophoben Phasen wird der Analyt gleich stark festgehalten, es ergeben sich ähnliche lnk-Werte. Diese Praxis sollte kritisch hinterfragt werden: Ohne auf die Eignung der (meist einfachen) verwendeten Analyte einzugehen, sollte man sich mit der prinzipiellen Frage auseinandersetzten, ob Retentionsfaktoren überhaupt ein geeignetes Vergleichskriterium darstellen: Das Retentionsverhalten (also ein Maß für die Hydrophobie) interessiert aus praktischer Sicht eher am Rande, denn: Bei vergleichenden Tests habe ich als Anwender eher das Ziel, Säulen zu finden, die bestimmte Analyte “ähnlich” (gut) trennen als Säulen zu finden an denen die Analyte nach ähnlicher Retentionszeit eluieren. Und ein Maß für die Trennfähigkeit ist nun mal die Selektivität (Trennfaktor, α). Ähnlich starke Wechselwirkungen - und somit ähnliche Retentionsfaktoren – bedeutet keines Falls auch ähnliches Selektivitätsverhalten der betreffenden Phasen. Dies hat sich bei diversen Untersuchungen immer wieder bestätigt, s. dazu beispielhaft Abb. 4. Dort werden die Retentions- (Balken) und Trennfaktoren (Linie) von trizyklischen Antidepressiva im sauren Acetonitril/Phosphatpuffer gezeigt. Bei recht unterschiedlichen Retentionsfaktoren abhängig vom hydrophoben Charakter der Phasen (unterschiedlich starke Wechselwirkungen) ergeben sich bis auf wenige Ausnahmen sehr ähnliche Trennfaktoren: Obwohl die Analyte unterschiedlich lang an den stationären Phasen festgehalten werden, zeigen diese ein ähnliches Selektivitätsverhalten. Ein solches wird vor allem bei neutralen oder - mithilfe des pH-Wertes - neutralisierten Molekülen beobachtet. In diesem Fall herrschen weitgehendst hydrophobe Wechselwirkungen zwischen den Analyten und der Oberfläche des Materials. Jene sind nicht besonders spezifisch, die individuellen Unterschiede der Phasen bzgl. Selektivität kommen nicht zum Tragen.

Abb. 4 zeigt dieses Phänomen bei der Trennung von Ketonen, polaren jedoch nichtionisierbaren Analyten. Und: Eine starke Wechselwirkung bedeutet nicht automatisch eine gute Selektivität, s. dazu auch Abb. 5, in der im Prinzip dieses Phänomen etwas anders dargestellt wird: Dort sind Retentionsfaktoren an diversen Säulen gegen die entsprechenden Trennfaktoren aufgetragen. Es ist so, dass die Peaks an den unterschiedlichen Säulen früh oder spät eluieren (gemäß „Hydrophobicity“ sind es Phasen mit unterschiedlichen Eigenschaften) aber das Selektivitätsverhalten dieser Säulen ist äußerst ähnlich: Alle α -Werte bewegen sich zwischen ca. α=1,1-1,2. Für eine Trenntechnik wie der HPLC dürfte Selektivität das wichtigste bzw. aussagefähigste Kriterium ist, wenn es um eine Überprüfung der Ähnlichkeit von RP-Phasen geht.


Um diese Annahme zu verifizieren, wurden mehrere chemometrische Analysen mit umfangreichem Datenmaterial (sehr unterschiedliche Trennbedingungen und unterschiedlichste Analyte) durchgeführt, s. dazu in Abb. 7 ein Beispiel aus diesen Untersuchungen. Je weiter entfernt sich die Variablen - hier Retentions- und Trennfaktoren - um den O-Punkt auf der y-Achse befinden, umso größer ist deren Einfluss auf die Differenzierung von Objekten, hier von Säulen. Wie leicht zu erkennen ist, sind dazu die α-Werte geeignet. Möchte man dennoch par tout k-Werte verwenden, eignen sich dafür höchstens “problematische” Substanzen, z. B. polare Analyte in ungepufferten Eluenten (z. B. k Cos, Coffein) oder starke Säuren in gepufferten Eluenten (k Phs, Phthalsäure und k Tes, Terephthalsäure) denn, nur wenn polare (ionische) Wechselwirkungen möglich sind, kommen die Unterschiede der Phasen zum Vorschein. Es sei denn, man erwartet ausschließlich hydrophobe Wechselwirkungen. In diesem Fall ist das Thema „Säulenvergleich“ und somit „Säulenauswahl“ allerdings sowieso nicht so brisant, jede „gute“ Säule kann verwendet werden, die Optimierung erfolgt sinnvollerweise eher über den Eluenten.
Abb. 7 (aus „Optimierung“, Abb. 18, S. 202)
5.5.2 Säulenvergleich, Vergleichskriterium: Ähnlichkeit von Selektivitäten
In [8] wird ausführlich auf diversifizierte Tests und Visualisierungstools zum Vergleich von Säulen eingegangen. Nachfolgend möchten wir kurz drei Tests vorstellen, die uns als recht aussagekräftig erscheinen. Als Zuordnungskriterium zum Vergleich von Säulen wird aus den weiter oben dargelegten Gründen die Selektivität (Trennfaktoren) verwendet.
- Selektivitätsplots
Hier werden Trennfaktoren gegeneinander aufgetragen, die bei Trennungen mit vermeintlich ähnlichen Phasen unter definierten chromatographischen Bedingungen ermittelt wurden. Je ähnlicher die Phasen sind, umso näher sollten sich die einzelnen Werte an die resultierende Diagonale anschmiegen. In einer Versuchsreihe wurden 68 unterschiedliche Analytpaare unter bewusst unterschiedlichen chromatographischen Bedingungen (9 unterschiedliche Eluenten) getrennt und die ermittelten α -Werte gegeneinander aufgetragen. Durch die große Anzahl der Werte und durch die unterschiedlichen experimentellen Bedingungen gelangt man zu recht gesicherten Aussagen bzgl. Ähnlichkeit der Phasen. Ein Beispiel für einen derartigen Plott stellt Abb. 8 dar. Hier werden Kromasil und LUNA (2) miteinander verglichen.

Abb. 8 Auftragung der Trennfaktoren von möglichst unterschiedlichen Analytpaaren an zwei Säulen, Erläuterungen, s. Text
Aus Abb. 8 kann zunächst entnommen werden, dass beide Säulen einen ähnlichen Charakter aufweisen. Dies wird auch so erwartet, denn bei Beiden handelt es sich um zwei hydrophobe, endcappte C18 -Phasen mit klassischer, hoher Belegung und auf Basis hochreinen Kieselgels. Auffallend ist der Ausreißer „21“. Der α -Wert entspricht der Trennung 2,2´-/4,4´-Bipyridyl nach einem Säulenbetrieb von 4 Wochen. Ein großer α -Wert ist ein Hinweis auf das Vorhandensein von Schwermetallionen in der Kiesegelmatrix, denn 2,2´-Bipyridyl ist ein Komplexbildner, 4,4´ dagegen nicht. Im Laufe des Säulenbetriebes reichern sich offensichtlich an der Kieselgel-Oberfläche von Kromasil mehr Metallionen an (Metallfritte?), die Neigung zur Komplexbildung nimmt zu. Oder aber diese Kiesegel-Charge von Kromasil enthält Spuren von Metallionen, die bei LUNA fehlen. Weiterhin fällt bei LUNA der etwas höhere hydrophobe Charakter auf, s. Trennung "37": Trennung von 3 Hydroxy-/4 Hydroxy-Benzoesäure, zwei schwachen Säuren. Tatsächlich weist LUNA einen etwas höheren Bedeckungsgrad im Vergleich zu Kromasil auf, die Phase verfügt über eine geringfügig größere hydrophobe Selektivität. Mit derartigen Auftragungen kann ermittelt werden, ob zwei vermeintlich ähnliche Säulen auch tatsächlich für diese Analyte ein ähnliches Selektivitätsverhalten aufweisen. Auch die „Batch-to-Batch“ Reproduzierbarkeit eines Materials kann hiermit getestet werden. Oder aber, ob eine bestimmte Säule in der Routine durch diese Probenmatrix und/oder diese chromatographischen Bedingungen (irreversibel) ihren Charakter verändert hat.
- Auftragung der Trennfaktoren von zwei recht ähnlichen Analytpaaren, die an den zu vergleichenden Säulen erhalten wurden, z. B. Phenol/Ethylbenzol und Phenol/Toluol.
Da diese Moleküle typische, einfache RP-Analyte und dazu sehr ähnlich sind (Differenz nur einer Methylgruppe zwischen Toluol und Ethylbenzol), sollten sie an unterschiedlichen Phasen mit ähnlichen Selektivitätsfaktoren zu trennen sein. Nur Phasen mit „wirklich“ anderem Charakter sollten hier „aus der Reihe tanzen“. Die drei Ausreißer in Abb. 9 stellen in der Tat „Sonderlinge“ dar: Gemini und XBridge sind Hybridmaterialien und Uptisphere UP 5MM1 ist die einzige Phase mit einer Phenyl- und einer Cyclopentenyl-Gruppe als Endgruppen. Mit diesem Test kann z. B. getestet werden, welche Säule(n) für welche Analyte ein gänzlich anderes Selektivitätsverhalten aufweist(en). Aus den Testergebnissen können auch geeignete Kandidaten für orthogonale Tests ermittelt werden.

Abb. 9 Auftragung der Trennfaktoren von zwei charakteristischen Analytpaaren an mehreren Säulen, Erläuterung, s. Text
- Auftragung der Trennfaktoren von zwei, mich interessierende Analytpaaren an unterschiedlichen Säulen.
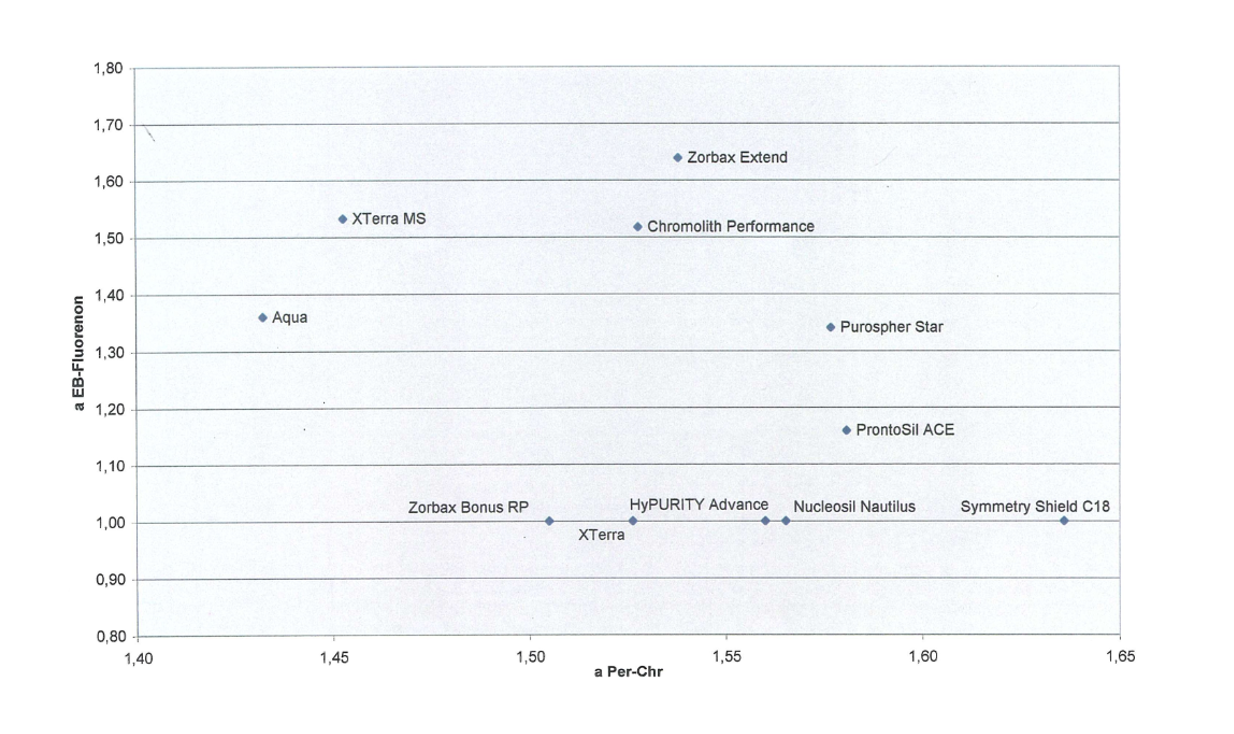
Es sollten solche Analytpaare gewählt werden, die die Ähnlichkeit der Phasen bzgl. Selektivität für eigene Fragestellungen offenbaren. Nehmen wir an, in einem Fall ist die Trennung sowohl von einfachen neutralen Molekülen als auch von Aromaten von Relevanz, s. dazu Abb. 10. Ethylbenzol/Fluorenon ist ein Maß für die hydrophobe Selektivität und Chrysen/Perylen für die aromatische Selektivität. XTerra MS und AQUA zeigen als recht hydrophobe Phasen eine gute hydrophobe Selektivität, wohingegen Symmetry Shield und Nautilus als polare Phasen eine gute aromatische Selektivität aufweisen. Möchte man nun sowohl kleine, neutrale als auch große, mehrkernige Aromaten trennen, wären Zorbax Extend, Chromolith und Purospher Star eine gute Wahl – die Platzierung einer Säule in der oberen rechten Ecke des α/α-Diagramms bedeutet eine gute Selektivität für beide Analytpaare. Mit diesem Test kann die Ähnlichkeit des von mir verwendeten Säulenportfolios für die von mir zu analysierenden Substanzen ermittelt werden.
5.5.3 Zwei einfache Tests zur Charakterisierung von RP-Phasen
Die soeben vorgestellten Tests liefern zweifelsohne hilfreiche und präzise Informationen, stellen doch einen Aufwand dar, der vermutlich in den wenigsten Labors zu vertreten wäre. Wir haben nun schon vor einigen Jahren anhand der Ergebnisse diverser Untersuchungen zwei einfache Tests entwickelt, die sich bewährt haben: Isokratische Läufe, kein Puffer, Retentionszeit unter 10 min - und dabei Gewinn von interessanten Informationen. Sie werden nachfolgend kurz vorgestellt:
Test 1
Eluent: 80/20 Methanol/Wasser (v/v, zuerst MeOH vorlegen)
Fluss: 1 ml/min
Temperatur: 35 °C
Detektion: UV, 254 nm
Inkektion von Ethylbenzol und Fluorenon, im Eluenten gelöst
Test 2
Eluent: 40/60 Methanol/Wasser (v/v, zuerst MeOH vorlegen)
Rest, wie oben
Injektion von Phenol, Benzylamin im Eluenten gelöst (frisch angesetzte Lösung!)
Ergebnisse und Kommentare
Test 1
Ergibt sich an einer Säule ein Trennfaktor größer ca. 1,4 handelt es sich um eine Phase mit einer guten hydrophoben Selektivität, ein Trennfaktor größer ca. 1,6 zeugt von einer Phase mit einer sehr guten hydrophoben Selektivität. Phasen mit einer guten hydrophoben Selektivität eignen sich für die Trennung kleiner, einkerniger Aromaten, für Homologen mit einem Unterschied in der Alkylkette, für Aldehyde, Ketone, Ester und für neutrale „unproblematische“ Analyte wie schwache organische Basen und schwache organische Säuren. Säulen, die hier kleine Trennfaktoren aufweisen, eignen sich für die Trennung kleinerer polaren Verunreinigungen, verdrillte Strukturen sowie Carotinoide, Steroide, Doppelbindungsisomere, mehrkernige Aromaten, ionisch vorliegende Analyte wie starke Säuren oder überhaupt Analyte mit polarem Charakter wie Phosphorlipide und Farbstoffe.
Test 2
Je später das Benzylamin im Vergleich zu Phenol eluiert, umso stärker ist der polare Charakter der Phase (genauer: Sie verfügt über Gruppierungen, die zu ionischen Wechselwirkungen in der Lage sind). Eine nahe Elution zu Phenol oder Koelution zeugt von einer hydrophoben Phase, eine Elution gar vor Phenol von einer sehr gut abgedeckten, sehr hydrophoben RP-Phase. Sollte eine derartige Phase für eine aktuelle Fragestellung die gewünschte Selektivität zeigen, wird man wohl mit dieser Säule in der Routine kaum ernsthafte Probleme bekommen: Gute Peakform, reproduzierbare Retentionszeiten, lange Lebensdauer.
5.6 Vorgehensweise bei der Methodenentwicklung in der Praxis
Die Auswahl der Trennsäule ist von entscheidender Bedeutung und steht daher an erster Stelle bei jeder Methodenentwicklung. In diesem Kapitel soll nun ein Leitfaden bei der Vorgehensweise gegeben werden mit dessen Hilfe es möglich sein sollte, schnell zu der am besten geeigneten Trennsäule zu kommen. Bevor wir jedoch zu der Liste kommen, sollten einige wichtige Regeln, die häufig missachtet werden, genannt werden, deren Einhaltung schneller zum Erfolg führt.
5.6.1 Das Zusammenspiel zwischen mobiler und stationärer Phase
Nur die Kombination aus mobiler und stationärer Phase (natürlich auch Temperatur) liefert die Trennung und manche Kombinationen sind nicht sinnvoll und können folglich entweder gar nicht berücksichtigt werden, oder nur dann (quasi aus Verzweiflung) heran gezogen werden, wenn alle anderen Kombinationen nicht funktionieren.
Die wichtigste Grundregel lautet dabei:
Verwenden Sie für die Chromatographie basischer Verbindungen bei unpolaren stationären Phasen (klassischen Umkehrphasen) bevorzugt aprotische Lösemittel (z. B. Acetonitril) als mobile Phasen und bei polaren stationären Phasen (Umkehrphasen mit eingebundenen polaren Gruppen) vorzugsweise protische Lösemittel (z. B. Methanol).
Warum ist das so?
Protische Lösemittel solvatisieren polare funktionale Gruppen und exponieren diese dadurch stärker. Die einzigen polaren funktionellen Gruppen, die man bei einer klassischen unpolaren Umkehrphase finden kann sind die Silanolgruppen. Werden diese durch Solvatisierung stärker hervorgehoben, so treten die ungewollten ionischen Wechselwirkungen mit den basischen Analyten stärker in den Vordergrund und die Elutionsbanden werden breit und asymmetrisch. Verwendet man bei diesen Säulen jedoch ein aprotisches Lösemittel wie Acetonitril, so kann das einsame Elektronenpaar am Acetonitril Wasserstoffbrücken mit den Silanolgruppen bilden und diese werden von den basischen Analyten abgeschirmt. Die Elutionsbanden sind deutlich schmäler und symmetrischer.
Bei polaren Umkehrphasen ist der Effekt genau anders herum. Hier werden die polaren Gruppen (meist Stickstoff haltige funktionelle Gruppen) durch protische Lösemittel exponiert und treten dabei mit den Silanolgruppen in Wechselwirkung. Dies hat den Vorteil, dass dadurch nicht der basische Analyt mit den Silanolgruppen in Wechselwirkung treten kann. Somit eluieren Basen bei der Verwendung von protischen Lösemitteln von polaren Umkehrphasen mit symmetrischen Elutionsbanden.
Bei der Trennung von sauren Analyten werden zur Erreichung der maximalen Selektivität möglichst starke Wechselwirkungen der Analyte mit den Silanolgruppen der stationären Phase benötigt. Folglich ist es hier sinnvoll auf klassischen, unpolaren stationären Phasen protische Lösemittel wie Methanol als mobile Phase einzusetzen, und auf polaren stationären Phasen die polaren Wechselwirkungen möglichst zu unterdrücken, und somit aprotische Lösemittel wie Acetonitril als mobile Phase zu nehmen.
Hat man sowohl Säuren als auch Basen in einer Probe vorliegen, so werden meist die für die Basen geeigneten mobilen Phasen verwendet, da breite und asymmetrische Elutionsbanden sich in der Regel schwerwiegender auf die analytische Fragestellung auswirken als eine etwas schlechtere Selektivität. Manchmal werden in solchen Fällen als Kompromiss auch Mischungen aus protischen und aprotischen Lösemitteln (Methanol und Actonitril) verwendet.
Ferner werden in der Umkehrphasen HPLC noch stationäre Phasen eingesetzt deren Oberfläche mit Phenylgruppen oder Pentfluorphenylgruppen modifiziert wurden. Diese funktionellen Gruppen können ihre maximale Selektivität nur dann ausspielen, wenn sie solvatisiert werden. Daher muss bei Gebrauch dieser Säulen zwingend ein protisches Lösemittel als mobile Phase verwendet werden. Die Verwendung von Acetonitril verbietet sich, da das einsame Elektronenpaar im Acetonitril mit der stationären Phase in Wechselwirkung tritt und somit die Wechselwirkung der Phenylgruppen mit dem Analyten zumindest unterdrückt, wenn nicht sogar ganz blockiert.
Hält man sich an diese nur wenigen Regeln, so kommt man schnell zum Erfolg.
5.6.2 Welche Trennsäulen sollten verwendet werden und wie setze ich diese ein?
Zunächst einmal müssen wir unterscheiden, ob wir alle unsere zu bestimmenden Analyte und unsere analytische Fragestellung kennen, oder ob es möglicherweise unbekannte Verunreinigungen gibt, die wir noch gar nicht kennen.
Kennen wir unsere Analyte und unsere Fragestellung, können wir die Säule zielgerichtet darauf auswählen. Wollen wir zum Beispiel organische Säuren trennen, die sich primär in der Kettenlänge des aliphatischen Restes, also in ihrer hydrophoben Selektivität unterscheiden, so probiert man am besten eine klassische, endcappte, hydrophobe Umkehrphase aus, denn diese bietet die für diese Fragestellung notwendige Methylengruppenselektivität. Wollen wir hingegen aromatische Carbonsäuren trennen, die sich in der Stellung der funktionellen Gruppen am aromatischen Ring unterscheiden, so wäre es nicht Ziel führend zunächst eine klassische Umkehrphase zu verwenden. Hier unterscheiden sich die Analyte vor allem in ihrer π-Elektronendichte am aromatischen Ring und solche Analyte können über π-π-Wechselwirkungen am effektivsten getrennt werden. Somit wäre in diesem Fall der Einsatz einer stationären Phase mit Phenylgruppen die erste Wahl. Wie bereits im Kap. 3 ausgeführt, wird in der HPLC die Trennung und damit die Auflösung primär über die Selektivität erreicht und diese wiederum hängt u A stark von der richtigen Trennsäule ab.
Wenn wir also Informationen zu den zu trennenden Analyten haben, können wir diese zur geschickten Auswahl der zu verwendeten Trennsäule nutzen.
Wenn wir die Heterogenität einer unbekannten Probe aufklären müssen, ist es notwendig, die Probe auf möglichst unterschiedlichen Trennsäulen mit orthogonaler Selektivität zu betrachten um auszuschließen, dass eine mögliche Koelution übersehen wird. Zur Auswahl dieser Trennsäulen orientiert man sich am Besten an den Kerneigenschaften der Trennsäulen, die unter Punkt 5 betrachtet wurden.
Daher sollte man zu Beginn zunächst einmal eine moderne C18 oder C8-Säule mit einem guten effizienten Endcapping ausprobieren. In den meisten Fällen ist man damit bereits erfolgreich. Zwei typische Vertreter wären z. B. hier: Chromolith High Resolution RP 18e von Merck und YMC Pro C8. Die Chromolith-Säule bietet den Vorteil der hohen Trenneffizienz bei relativ geringem Gegendruck und gleichzeitig geringer Säulenlänge. C8-Säulen sind von ihrem chromatographischen Verhalten gegenüber C18-Säulen vorzuziehen, da sie in der Regel eine höhere Belegungsdichte besitzen und von daher weniger unerwünschte Sekundärwechselwirkungen zwischen Basen und stationärer Phase möglich sind. Werden in unserer Probe also etwas stärkere Basen erwartet, so ist eine partikuläre C8-Säule der monolithischen C18-Säule vorzuziehen. Eine monolithische C8-Säule wäre zu nieder-retentiv und ist daher für den allgemeinen Einsatz keine Option, in Spezialfällen jedoch durchaus eine Alternative.
Sollte die Trennung auf einer klassischen Umkehrphase nicht funktionieren, so muss grundlegend der Typ an stationärer Phase gewechselt werden. Nur in Einzelfällen ist der Wechsel zu einer klassischen Umkehrphase eines anderen Herstellers Ziel führend. Die Säule mit der man nach der klassischen Umkehrphase den größten Erfolg hat ist eine Umkehrphase mit einer eingebundenen polaren Gruppe. Als typische Vertreter hierfür wären zu nennen, ProntoSIL C18 ace-EPS von Bischoff oder Symmetry Shield RP 18 von Waters. Sie zeigen vergleichbare hydrophobe Retention wie klassische Umkehrphasen (die meisten Umkehrphasen mit eingebundener polarer Gruppe sind nieder-retentiv) und weisen eine recht hohe polare Selektivität auf.
Sollte auch diese Säule nicht die gewünschte Trennung ergeben, wäre die nächste Wahl eine Phenyl-Säule. Beispiele für diesen Typ Trennsäule bieten XSelect CSH Phenyl-Hexyl von Waters oder Zorbax Phenyl von Agilent Technologies. Viele Hersteller nehmen als Spacer für die Phenylgruppe Hexyl. Mit zunehmender Kettenlänge wird die π-π-Wechselwirkung stärker [16].
Sollte auch diese Trennsäule nicht funktionieren, wäre eine klassische Umkehrphase mit hohem Metallgehalt und ohne Endcapping bzw. eine moderne C30-Säule ohne Endcapping und mit hoher molekularer Formerkennung (shape selectivity) die Säule der Wahl. Empfehlungen aus dieser Kategorie sind: LiChrospher RP 18 von Merck oder ProntoSIL 200 C30 von Bischoff. Ein Praxistipp zur „shape selectivity“ an dieser Stelle: Die molekulare Formerkennung ist stark Temperaturabhängig. Je niedriger die Säulentemperatur umso höher die „shape selectivity“. Daher sollten „shape“-selektive Trennungen am besten bei niedrigen Temperaturen erfolgen.
Eine weitere Option wäre der Einsatz einer Cyano – Säule (CN). Eine typische klassische CN-Säule wäre Nucleosil CN von Macherey Nagel oder LUNA CN von Phenomenex. Diese Säulen trennen über Dipolwechselwirkungen und somit bevorzugt Verbindungen, die sich in ihrem Dipolmoment unterscheiden. Aufgrund der niedrigen hydrophoben Selektivität kommt es auf diesen Säulen jedoch häufig zu Koelutionen von Verbindungen, die ähnliche Dipolmomente besitzen und sich nur wenig in der Hydrophobie unterscheiden. Dennoch bieten diese Säulen eine interessante Alternative für Gruppentrennungen, die dann in Kombination mit 2D-LC bzw. POPLC ihre Stärken ausspielen kann.
Sehr hydrophobe Analyte treten häufig sehr stark mit der stationären Phase in Wechselwirkung und werden von konventionellen Umkehrphasen manchmal nicht mehr eluiert. Hier hilft der Einsatz einer sehr kurzkettigen Alkylphase, die lediglich mit Trimethylsilylgruppen modifiziert wurde. Eine bewährte Säule hierfür ist z.B. Zorbax TMS von Agilent Technologies.
Bei der Auswahl der oben genannten Säulen kann es dazu kommen, dass auf manchen Trennsäulen zwei Komponenten getrennt werden, dafür aber andere Komponenten, die auf andern Trennsäulen getrennt werden plötzlich zusammenfallen. In solchen Fällen bietet sich die Verwendung der Phasen optimierten Flüssigkeitschromatographie (POPLC) an [17, 18]. Das Prinzip ist kurz in der Abbildung weiter unten dargestellt. In einem Säulenkit befindet sich ein Set aus segmentierten Trennsäulen. Dieses Set bildet die oben beschriebenen unterschiedlich selektiven Trennsäulen ab. In einer jeweiligen Basismessung auf den einzelnen Trennsäulen wird die Retentionszeit einer jeden Verbindung bestimmt, in eine Software eingegeben und mit deren Hilfe die ideale Zusammensetzung der Trennsäule bestimmt. Die POPLC kann durch die zielgerichtete Kopplung die „beste“ Säulenzusammensetzung für die aktuelle Trennung liefern. Übersteigt die Summe der zu trennenden Analyte jedoch eine Anzahl von ca. 20, so bringt meist die POPLC-Optimierung auch keine Lösung. Hier hilft dann nur noch hohe Peakkapazität (also 2-D-HPLC) bzw. hohe Bodenzahl.

5.6.3 Was tun, wenn die Analyten sehr polar sind und auf den oben genannten Säulen keine Retention aufweisen?
Polare Analyten stellen sehr häufig eine große Herausforderung an die Umkehrphasenchromatographie dar. Denn selbst mit sehr polaren (rein wässrigen) mobilen Phasen kann man nur wenig oder gar keine Retention erhalten. Für diese Fälle gibt es bestimmte Strategien, die im Folgenden besprochen werden sollen.
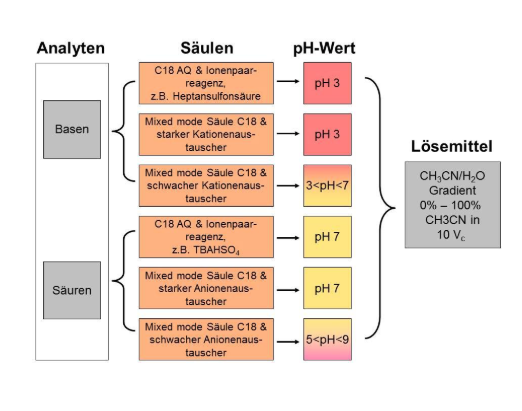
5.6.3.1 AQ-Säulen, polare RP-Säulen und Ionenpaarchromatographie
Sollen polare Analyte in der Umkehrphasen HPLC getrennt werden, müssen sehr polare mobile Phasen verwendet werden. Denn erhöht man den Wassergehalt der mobilen Phase, so steigt die Retention. Bei klassischen Umkehrphasen ist dies (zumindest bei Gegendrücken unter 200 bar) nur bis zu einem Anteil von organischem Lösemittel größer 5% der Fall. Wird dieser unterschritten bricht die Retention schlagartig zusammen. Klassische C18 Säulen können also nicht mit 100% wässrigen Eluenten betrieben werden. Der Grund liegt in der hohen Oberflächenspannung des Wassers. Über 99% der spezifischen Oberfläche der stationären Phase befindet sich in den Poren. Unterschreitet der Gehalt des organischen Lösemittels in der stationären Phase 5%, so bildet sich am Eingang der Poren ein „Tropfen“, der den Austausch der Analyte in und aus den Poren verhindert. Erst bei einem Gegendruck über 200 bar ist der Austausch wieder möglich. [7]. Daher wurden stationäre Phasen entwickelt, mit denen im Umkehrphasenmodus in rein wässrigen Medien gearbeitet werden kann. Dazu zählen so genannte AQ-Säulen. Diese tragen häufig AQ im Namen oder heißen Aqua oder Hydro. Manchmal werden deren Namen aber auch nur mit Wasser assoziiert, wie z.B. Atlantis. Prinzipiell kann die Wasserverträglichkeit auf dreierlei Art erreicht werden:
a) Unterbelegung: z. B. Platinum C18 von Alltech oder Atlantis T3 von Waters. Diese Säulen sind klassische C18-Säulen haben aber eine deutlich geringere Belegungsdichte als konventionelle C18-Säulen. Sie haben ein konventionelles Endcapping und weisen auch die Selektivität wie konventionelle Säulen auf. Durch die Unterbelegung wird jedoch eine bessere Zugänglichkeit der Oberfläche erreicht, was dazu führt, dass auch polare Restsilanolgruppen „gesehen“ und solvatisiert werden können und dadurch die Porenzugänglichkeit erhalten bleibt.
b) Polares Endcapping: z.B. Aquasil C18 von Thermo oder ReproSIL C18 AQ von Dr. Maisch. Bei diesen stationären Phasen wird statt eines konventionellen Endcappings mit Trimethylsilylgruppen ein Endcapping mit einem kleinen Silan durchgeführt, welches eine polare Gruppe enthält. Diese sorgt für die Benetzung der Oberfläche und somit die Zugänglichkeit der Poren.
c) Umkehrphasen mit eingebundener polarer Gruppe: z.B. Ascentis RP Amid von Sigma Aldrich oder XBridge Shield RP 18 von Waters. Diese stationären Phasen besitzen eine polare Gruppe (Amid, Carbamat oder Harnstoff), die in die Alkylkette eingebaut ist. Das Endcapping dieser Materialien ist konventionell (Trimethylsilylgruppen). Bei diesen Säulen sorgt die in die Alkylkette eingebaute polare Gruppe für die Benetzung der Oberfläche.
Schließlich kann auch an weitere sehr polare Phasen gedacht werden wie diversen fluorierten Phasen oder auch Zorbax Bonus RP bzw. Synergi POLAR RP. Eine ungewöhnliche dennoch immer wieder erfolgreiche Massnahme lautet: Typischer RP-Eluent (z. B. Puffer/ACN) plus Kieselgel, das ist die aus den 1980er Jahre bekannte, wässrige Normalphasenchromatographie („Aqueous Normal Phase“, im Gradientenmodus heißt dies heute modern HILIC, s. weiter unten).
Sind die zu trennenden Analyte so polar, dass selbst 100% wässrige mobile Phasen nicht ausreichen, die Analyte auf den oben genannten Trennsäulen auf Retention zu bringen, so hilft zumindest für ionisierbare Analyte die Ionenpaarchromatographie. Bei der Ionenpaarchromatographie setzt man der mobilen Phase zur Probe entgegengesetzt geladene Reagenzien zu, die eine polarer Gruppe und einen unpolaren Rest enthalten. Diese zugesetzten anionischen bzw. kationischen Tenside bilden dann mit den polaren kationischen bzw. anionischen Analyten ein so genanntes Ionenpaar, das dann nach außen elektroneutral ist und so auf Umkehrphasen retardiert werden kann. Andere Vorstellungen gehen davon aus, dass die Oberfläche der stationären Phase mit dem entsprechenden Ionenpaarreagenz belegt wird und so ein Ionenaustauscher vorliegt, auf dem es dann möglich ist, die entgegengesetzt geladenen Analytionen zu trennen. Die Wahrheit liegt wohl zwischen diesen beiden Modellen. Ionenpaarchromatographie ist eine sehr elegante Möglichkeit ionische Analyten im Umkehrphasenmodus zu chromatographieren, jedoch sind in der Praxis einige Dinge zu beachten:
a) Die Gleichgewichtseinstellung eines Ionenpaarreagenzes mit der stationären Phase dauert sehr lange. Die Säule sollte über Nacht bei kleinem Fluss äquilibriert werden. Am Besten eignen sich AQ-Säulen zur Verwendung in der Ionenpaarchromatographie, denn diese haben eine gute Zugänglichkeit der kompletten chromatographischen Oberfläche und sind generell um einen Faktor zwei schneller im Gleichgewicht als konventionelle C18-Säulen (das gilt übrigens auch für den klassischen Einsatz dieser Säulen außerhalb des polaren Einsatzbereiches).
b) Bei der Verwendung eines Ionenpaarreagenzes muss stets dafür Sorge getragen werden, dass sowohl der Analyt als auch das Ionenpaarreagenz in dissoziierter Form vorliegt. Daher ist es notwendig, die Eluenten auch entsprechend abzupuffern.
c) Ionenpaarreagenzien sind auch immer Tenside und sind daher nur sehr schwer in reiner Form kommerziell erhältlich. Daher finden sich häufig Verunreinigungen in den Ionenpaarreagenzien, die im Falle einer Gradientelution zunächst am Säulenkopf angereichert werden, um dann bei steigender Lösemittelstärke von der Trennsäule eluiert zu werden und als so genannte „Geisterpeaks“ im Chromatogramm aufzutauchen. Kommt es zu keiner Koelution mit den zu bestimmenden Analyten ist das nicht schlimm. Es ist jedoch notwendig die Geisterpeaks im Chromatogramm zu lokalisieren und zu kennen. Daher ist es unbedingt notwendig eine Blindinjektion vor dem eigentlichen chromatographischen Lauf durchzuführen und auch die Äquilibrierungszeit zwischen den einzelnen chromatographischen Läufen konstant zu halten, da sonst bei längerer Äquilibrierung sich die Intensität der „Geisterpeaks“ ändert.
d) Die erfahrungsgemäß reinsten Ionenpaarreagenzien sind: zur Trennung von Anionen Tetrabutylammoniumhydrogensulfat (TBAHSO4) und zur Trennung von Kationen Natriumdodecylsulfat (SDS). Typische eingesetzte Konzentrationen in den Eluenten sind 10 mM Ionenpaarreagenz und 20 mM Puffer.
e) Da Ionenpaarreagenzien nicht verdampfbar sind, verbietet sich der Einsatz von massenspektrometrischen Detektoren und Aerosoldetektoren. Es besteht jedoch die Möglichkeit, das Eluat mittels Supressoren vor dem Detektor von den Ionenpaarreagenzien zu befreien und dann dennoch diese Detektoren zu verwenden [19].
Eine weitere elegante Möglichkeit die Ionenpaarchromatographie zu umgehen stellt die Verwendung so genannter „mixed mode“ Säulen dar.
5.6.3.2 Mixed Mode Säulen
Mixed mode Säulen enthalten Umkehr- sowie diverse andere funktionelle Gruppen, so z. B. oft komplexfähige oder Ionenaustauscher-gruppen. Dadurch, dass hier die austauschfähigen Gruppen kovalent auf die stationäre Phase aufgebunden sind, bieten diese Materialien eine elegante Möglichkeit ionisierbare polare Analyte ohne den Zusatz eines Ionenpaarreagenzes zu retardieren. Der mobilen Phase können bei der Verwendung von mixed mode Säulen flüchtige Puffer zugesetzt werden, so dass auch Aerosoldetektoren und massenspektrometrische Detektoren verwendet werden können. Es gibt einige Säulenhersteller, die sich auf mixed mode Säulen spezialisiert haben. Häufig eingesetzte mixed mode Säulen sind die Primesep-Trennsäulen der Firma Sielc. Diese sind als starke Kationenaustauscher (Primesep 100), schwache Kationenaustauscher (Primesep 200), starke Anionenaustauscher (Primesep B) und schwache Anionenaustauscher (Primesep B2) erhältlich, so dass zu jeder analytischen Aufgabenstellung auch die entsprechende Applikation zur Verfügung steht. Eine Säule mit einer komplexfähigen Gruppe am Alkylrest wäre Primesep C. Als weitere Beispiele vom Mixed Mode Säulen seien Acclaim Mixed Mode HILIC 1, Scherzo SM-C18 und Obelisc R oder N genannt. Weitere Säulen mit mehreren Ionenaustauschergruppen und einem zusätzlichen Heteroatom wie S erscheinen uns als zu speziell und finden hier nicht weiter berücksichtigt. Der besondere Charme dieser Säulen in der Praxis liegt darin, dass durch die Erhöhung des Acetonitrilgehaltes während des Gradienten nicht nur die Lösemittelstärke für den Umkehrphasenretentionsmechanismus erhöht wird, sondern gleichzeitig das Protolysegleichgewicht der ionenaustauschfähigen Gruppen zurückgedrängt wird, was zu einer Elution der ionischen Analyten führt.
Streng genommen stellt jede Kieselgel basierende Umkehrphase auch eine mixed mode Säule dar, denn sie besitzt neben den Alkylketten auch Silanolgruppen, die schwache kationenaustauschfähige Gruppen darstellen. Einige C18-Materialien der 1. Generation, wie Partisil ODS von Whatman oder Spherisorb ODS1 von Waters haben lediglich sehr kleine Kohlenstoffgehalte (kleiner 5%) und eine hohe Anzahl freier Silanolgruppen. Auf diesen Säulen lassen sich sehr gut stark basische Verbindungen trennen und auch mit symmetrischen Peakformen eluieren. Das liegt daran, dass die Silanolgruppen in einer so hohen Anzahl bei diesen stationären Phasen vorhanden sind, dass sie nicht überladen werden und daher symmetrische Elutionsbanden resultieren.
In den Abschnitten zuvor haben wir behandelt welche Möglichkeiten es gibt ionisierbare polare Analyte zu retardieren. Im Folgenden zeigen wir nun Möglichkeiten auf, auch neutrale Analyte neben ionisierbaren Analyten zu retardieren und zu chromatographieren.
5.6.3.3 Ionenausschlusssäulen/Ligandenaustauschchromatographie
Bei diesen Materialien handelt es sich um polymerbasierende stationäre Phasen, genauer erschöpfend sulfonierte Polystyrol/Divinylbenzol Copolymere. Der Retentionsmechanismus dieser Trennungen ist sehr komplex und liegt häufig in der Möglichkeit begründet, dass die Analyte mit dem Kation der stationären Phase Komplexliganden ähnliche Wechselwirkungen auszuüben [20]. Die Trennungen funktionieren jedoch häufig sehr gut v.a. für polare Analyte, die viele Hydroxylgruppen tragen, wie beispielsweise Zucker. Als mobile Phase wird dabei leicht angesäuertes Wasser verwendet. Die Trennsäulen gibt es nicht nur in der sauren H+-Form, sondern auch mit verschiedenen Metallen (Ca2+ und Pb2+) belegt. Dadurch ergeben sich unterschiedliche Selektivitäten. Für die Trennung von Zuckern werden primär die Metall-belegten Trennsäulen verwendet, während für die Trennung von organischen Säuren die Verwendung der H+-Form bevorzugt ist. Diese Trennsäulen gibt es von vielen Herstellern. Als dafür bewährte Trennsäule wäre Shodex K-801 zu nennen.
5.6.3.4 HILIC (hydrophilic interaction liquid chromatography)
Bei der HILIC werden polare stationäre Phasen mit mobilen Phasen betrieben, die mindestens aus einem polaren und einem unpolareren Lösemittel bestehen. Meist sind es Acetonitril/Wasser Mischungen mit hohem Acetonitril-Anteil. Es handelt sich also bei der HILIC um eine wässrige Normalphasenchromatographie [21, 22]. Wie in der Abbildung dargestellt reichert sich der polarere Teil der mobilen Phase an der polaren stationären Phase an. Die polareren Analyte bevorzugen das Verweilen nahe der polaren Oberfläche und werden daher stärker retardiert.

In der Praxis ist das Arbeiten mit HILIC völlig anders als das in der Umkehrphasenchromatographie und so kommt es häufig vor, dass Anwender die gewohnt sind mit Umkehrphasenchromatographie zu arbeiten beim Arbeiten mit HILIC Schiffbruch erleiden. Wenn Sie die folgenden Punkte beachten gelangen Sie schneller zum Erfolg:
a) Die Gleichgewichtseinstellung bei HILIC dauert deutlich länger als bei RP-Trennungen, in der Regel doppelt so lange. Reicht es in der Umkehrphasenchromatographie aus, die Säule nach einem Gradientlauf 5 bis 10 Säulenvolumina äquilibrieren zu lassen, so muss in der HILIC 10 bis 20 Säulenvolumina nach einem Gradientlauf äquilibriert werden.
b) Der Gradientverlauf in der RP-Chromatographie überstreicht in der Regel fast den kompletten Polaritätsbereich von Wasser zu Acetonitril. Bei HILIC wird der Gradient lediglich von 95% Acetonitril zu maximal 60% (meist sogar nur 70%) Acetonitril gefahren. Dabei beträgt das Gradientvolumen in Analogie zur Umkehrphasenchromatographie 5 bis 15 Säulenvolumina.
c) Die Beladbarkeit von HILIC-Säulen ist häufig deutlich geringer als jene von RP-Säulen. Auch dies ist zu beachten.
d) Der Puffer und v.a. die Pufferkonzentration hat einen stärkeren Einfluss auf die Trennung als in der Umkehrphasenchromatographie. Sie hat einen direkten Einfluss auf die Dicke der Schicht der polareren mobilen Phase („pseudostationären Phase“) nahe der Oberfläche. Auch hier ist auf robuste Verhältnisse zu achten.
Trotz dieser Einschränkungen stellt HILIC eine elegante Möglichkeit dar, polare Analyte zu retardieren und zu trennen.
In der HILIC werden verschiedene polare stationäre Phasen eingesetzt, die unterschiedliche Selektivität besitzen. Prinzipiell muss man zwischen drei unterschiedlich selektiven Typen unterscheiden: schwache Anionenaustauscher (mit Aminopropyl-gruppen modifiziertes Kieselgel) und Amidsäulen, schwache Kationenaustauscher (meist unbelegtes (nacktes) Kieselgel) und neutrale Träger (Diol oder zwitterionische stationäre Phasen (ZIC-HILIC)). Bei ionisierbaren Verbindungen kann es zu dem eigentlichen Verteilungsgleichgewicht zwischen den polareren Oberflächen nahen mobilen Phase („pseudostationären Phase“) und der weniger polaren mobilen Phase zusätzlich zu ionischen Wechselwirkungen kommen, die dann unterschiedliche Trenneigenschaften auf den unterschiedlichen stationären Phasen zur Folge haben. Bei den elektroneutralen stationären Phasen haben die zitterionischen Träger, bei denen auf der Oberfläche ein Betain aufgebunden ist gegenüber den Diol-Trägern den Vorteil, dass bei gleicher mobiler Phase die Retention der stationären Phase deutlich höher ist. Das liegt daran, dass die Oberflächen nahe polarere mobile Phase durch die ionischen Gruppen eine dickere Schicht ausbilden kann und somit die Retention für die polaren Analyte steigt. Die Selektivität der zwitterionischen stationären Phasen ist ähnlich zu der von Diol-Phasen.
Daher empfiehlt es sich beim praktischen Arbeiten neutrale, anionische und kationische polare stationäre Phase hinsichtlich Ihrer Selektivität zu prüfen.
5.6.3.5 Poröser Kohlenstoff
Poröser Kohlenstoff als stationäre Phase wurde in den frühen 1980er Jahren von John Knox und Mitarbeitern entwickelt und erforscht [23]. Kommerziell sind diese Trennsäulen bei der Firma Thermo unter dem Handelsnamen Hypercarb erhältlich. Alternativ dazu gibt es von der Firma Zirchrom ein mit Kohlenstoff beschichtetes Zirkoniumdioxidmaterial ähnlicher Selektivität. Der Vorteil des Grafitmaterials besteht darin, dass bestimmte polare organische Moleküle, besonders starke Wechselwirkungen mit dem Kohlenstoff eingehen können und somit eine sehr hohe Retention aufweisen. Dies liegt primär daran, dass einsame Elektronenpaare der Analyte, wie sie z. B. bei Aminogruppen, Hydroxylgruppen oder Carbonylgruppen vorliegen, in Wechselwirkung mit der Elektronenwolke des leitenden Graphits treten können. Darauf beruht auch eine hohe Selektivität zur Trennung von Isomeren die π-Elektronen enthalten. Da diese π-Elektronen in den Analyten mehr oder minder stark exponiert vorliegen kommt es zu mehr oder minder starken Wechselwirkungen mit der stationären Phase und somit zu hoher Selektivität.
Selbst sehr polare Analyte wie underivatisierte Aminosäuren, Oligosacharide oder Nucleinbasen können auf diesen Säulen retardiert werden.
Ein weiterer Vorteil ist die hohe Temperatur und pH- Beständigkeit der Kohlenstoffmaterialien.
Der Nachteil der Kohlenstoffsäulen besteht in einer im Vergleich zu Kieselgel schlechteren Porengrößenverteilung, was in der Praxis leicht asymmetrische, in den unteren 10% der Peakhöhe tailende Peakformen zur Folge hat.
5.7 Säulenscreening
Bei einer neuen Methodenentwicklung sollte möglichst schnell eine geeignete Trennsäule gefunden werden. Die effizienteste und schnellste Vorgehensweise dahin liegt in einem Säulenscreening, in dem die oben beschriebenen möglichst orthogonalen stationären Phasen in einem Screening abgefahren werden und einfach die Anzahl der Komponenten ermittelt wird die angetrennt werden kann. Die stationäre Phase, die die beste Trennung (größte Anzahl an Peaks) liefert kann dann zur Feinoptimierung herangezogen werden.
Damit dieses Säulenscreening möglichst schnell und effektiv ablaufen kann, wird in der Regel ein Säulenwechsler verwendet, in den bis zu 6 verschiedene Tennsäulen in den Säulenofen eingebaut werden können. Als Säulendimension für den Wechsler empfiehlt es sich möglichst kurze Trennsäulen (Länge: 50 bis 100 mm) mit Innendurchmessern von 3.0 bis 4.6 mm zu verwenden. Kleinere Innendurchmesser wären für diese Screenings ungeeignet, da das Verhältnis Volumen der Säule zu Volumen der Apparatur den Faktor 10 unterschreiten würde. Übersteigt das Apparaturvolumen 10% des Säulenvolumens, so kommt es zu einer deutlichen Bandenverbreiterung der Peaks außerhalb der Trennsäule, was in der praktischen Konsequenz zu einer Peakverbreiterung führt. Das bedeutet, dass die Trennung, die in der Trennsäule statt fand gleich wieder durch die Bandenverbreiterung zu Nichte gemacht wird.
Als mobile Phase sollte beim Säulenscreening der komplette Polaritätsbereich abgedeckt werden. Dies bedeutet bei klassischen Umkehrphasen, dass ein Gradient von 10-20% bis 95% Acetonitril abgedeckt wird (zum Gradienten s. auch Kap. 4). Die Steilheit des Gradienten sollte ca. 10 Säulenvolumina betragen. Bei polaren stationären Phasen muss Methanol statt Acetonitril oder eine Mischung aus Beiden verwendet werden. Ferner ist es notwendig bei dem Screening verschiedene pH-Werte zu betrachten, damit etwaige Selektivitätsänderungen, die von dem pH-Wert herrühren erkannt werden. Daher sollte bei pH 2, pH 7 und falls zu erwarten ist, dass basische Komponenten vorhanden sind auch pH 9 zum Screening herangezogen werden. An dieser Stelle sei noch mal darauf hingewiesen, dass beim Arbeiten über pH 7 Vorsicht geboten ist (siehe Abschnitt 4: pH-Stabilität). Die Säulen sollten beim Screening bei einer Lineargeschwindigkeit von ca. 3 mm/s (das entspricht bei einer ID 4.6 mm Säule ca. 1,5 ml/min, bei einer I.D. 3 mm Säule ca. 0,7 ml/min) betrieben werden. Die Steilheit des Gradienten soll etwa 10 Säulenvolumina betragen. Das bedeutet bei der 50 mm langen Säule wird der Gradient in ca. 4 Minuten und auf der 100 mm langen Säule in ca. 8 Minuten durchlaufen. Mit diesem Versuchsdesign ist es möglich ein Säulen- und Eluenten-Screening über Nacht durchlaufen zu können.
Die effektive Vorgehensweise bei der Methodenentwicklung wird in den folgen Grafiken nochmals zusammenfassend illustriert:
Vorgehensweise bei klassischer Umkehrphasenchromatographie
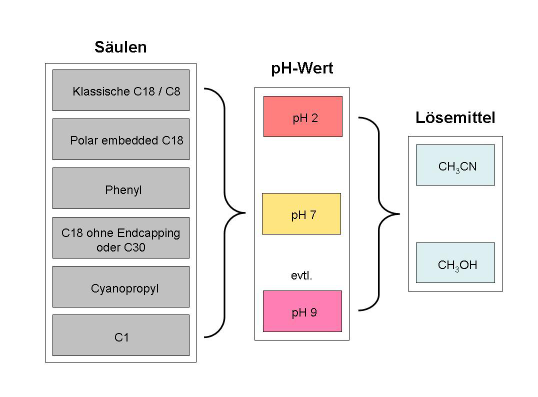
Vorgehensweise bei polaren Analyten
Vorgehensweise bei Ionenpaarchromatographie oder der Verwendung von mixed mode Säulen:

Vorgehensweise bei HILIC

Abschließend möchten wir Ihnen einige Beispiele von geeigneten Säulenportfolios abhängig von der Fragenstellung bzw. eventuellen Vorlieben für einen Hersteller vorstellen. Die angegebenen Säulen sind keinesfalls als Empfehlungen zu verstehen, vielmehr stellen sie Beispiele für eine bestimmte Intention dar. Selbstverständlich kommen auch Alternativsäulen, die Sie mithilfe von Datenbanken (s. weiter unten) finden können, in Frage.
Erläuterung: Je nachdem befindet sich in den zwei Tabellen entweder der Typ einer stationären Phase (z. B. „Biphenyl“ oder „PFP“) oder aber der Name einer konkreten Säule. Im ersten Fall können Sie je nach Verfügbarkeit und Lieferantenvorlieben entscheiden, welche Bi(Di)phenyl- oder welche Pentafluorphenylsäule Sie einsetzen möchten. Im zweiten Fall wird der Name der Säule als typischen Vertreter für diesen charakteristischen Typ des Materials genannt.

Kurzkommentare zu Tabelle 1
Spalte 1: Angenommen, Sie möchten gerne Säulen von Waters verwenden, da Waters Ihr Säulenhauptlieferant ist. Sie haben in diesem fiktiven Beispiel eine völlig unbekannte Probe und Sie würden gerne für den Nachtlauf mithilfe des Säulenwechslers recht unterschiedliche Säulen testen, also Sie brauchen Divergenz an Waters-Säulen.
Spalte 2: Angenommen, Sie vermuten in Ihrer Probe starke Basen und Sie hätten gerne Säulen getestet, die im Alkalischen stabil sind.
Spalte 3 und 4 sind selbsterklärend.
Spalte 5: Diese Säulen eignen sich für den Fall, dass Sie in Ihrer Probe Substanzen mit völlig unterschiedlichem Charakter erwarten/vermuten.
Spalte 6: Die Situation: Sie möchten gerne Säulen von Sigma Aldrich verwenden und Sie wissen, Sie haben wahrscheinlich polare Analyte, also bräuchten Sie die „ganze“ Vielfalt an polaren Phasen von Sigma Aldrich.
Nach dem Finden der „besten“ Säule über Nacht wäre nach diesem Test entsprechend unserem Kommentar ein weiterer Lauf sinnvoll: „Sicher“ ist sicher und ein orthogonaler Test im Falle von unbekannter Proben liefert parallel zu einer DAD-LC/MS-MS-Kopplung eine weitere Sicherheit in punkto Peakhomogenität.

Kommentare zu Tabelle 2
Spalte 1 bis 3 sind selbsterklärend.
Spalte 4: Bei 2D-Experimenten sind orthogonale Selektivitäten essentiell. Für die angegebenen Säulentypen dürfte die Auswahl einer geeigneten mobilen Phase keine größeren Probleme bereiten.
Spalte 5: Neben Beispielen von moderneren Säulentypen mit diversen Kombinationen an Funktionalitäten, haben wir auch Zorbax ODS aufgenommen. Wie z. B. LiChrospher stellt dieser alte Klassiker eine stationäre Phase dar, die nahezu „alle“ Arten von Wechselwirkungen ermöglicht, die für eine gute Selektivität wichtig sein können. Von hydrophoben Wechselwirkungen (ca. 17% C) über Ionenaustausch, Wasserstoffbrückenbindungen, Dipol-Wechselwirkungen usw. bis hin zur Komplexbildung. Das Material verfügt nämlich u A über freie, geminale und vicinale Silanolgruppen, Siloxanbindungen bis hin zu Metallionen als Kontaminationen.
Spalte 6 enthält schließlich Beispiele von Materialien, die einen recht unterschiedlichen Charakter aufweisen; wir haben bewusst auch zwei völlig unterschiedliche C18-Säulen berücksichtigt. Zum Kommentar: Je ähnlicher die Moleküle chemisch sind (auch ähnliche Polarität) umso wichtiger wird die Verwendung von Methanol und das Chromatographieren bei niedrigen Temperaturen. Die langsame Kinetik verbietet darüber hinaus höhere Flüsse.
5.8 Säulendatenbanken
Oft hat man eine gute Trennsäule, diese ist aber aus irgendwelchen Gründen nicht lieferbar oder deren Produktion wurde gar eingestellt oder die neuen Produktionschargen der Säulen zeigen eine völlig andere Selektivität und die Trennung funktioniert plötzlich nicht mehr. Es kann aber auch passieren, dass Sie mittels einer möglichst orthogonalen HPLC-Methode die Richtigkeit der ersten Methode beweisen müssen oder bei einer Methodenentwicklung eine Koelution haben. All das kennen wir aus dem Laboralltag. Hier tritt dann die Frage auf: Wie finde ich die zu meiner Trennsäule ähnlichste oder unähnlichste Trennsäule?
Die Antwort liegt wie heute nur all zu oft im Internet. Unter:
http://www.usp.org/app/USPNF/columns.html
liegen zwei Säulendatenbanken. In den Datenbanken sind Daten von Hunderten von HPLC-Säulen hinterlegt, die mit Hilfe chromatographischer Tests, so wie sie in Absatz 5 beschrieben sind, charakterisiert wurden [24-27]. Die erste Datenbank basiert auf dem USP Test von NIST („National Institut of Standards and Technology“) zur Charakterisierung von Umkehrphasen. Leider ist dieser Test nicht sehr präzise. In der zweiten Datenbank der PQRI Datenbank mit ca. 590 Säulen wurden zur Ermittlung der Daten jeweils fünf chromatographische Tests bei unterschiedlichen pH-Werten durchgeführt, so dass mit dieser Datenbank ein sehr guter Selektivitätsvergleich der stationären Phasen auch in Abhängigkeit zu den entsprechenden pH-Werten der mobilen Phasen möglich ist. Die unterschiedlichen Säulenparameter werden je nach pH-Wert der mobilen Phase und der Tatsache ob saure oder basische Analyte in der Probe zugegen sind entsprechend gewichtet. Es wäre ja z. B. unnötig die Ionenaustauschkapazität zweier Säulen bei der Berechnung des Fittingfaktors mit ins Kalkül zu ziehen, wenn die Probe gar keine basischen Analyte besitzt. So erhält man bei jedem Vergleich zweier Säulen einen Fittingfaktor, der ein direktes Maß über die Güte der Übereinstimmung der Säulen ist. Es lassen sich für jede Methode einfach die ähnlichsten, aber auch wie man in der Abbildung erkennt, die zur ausgewählten Säule unterschiedlichste Trennsäulen ermitteln. Der Vollständigkeit halber sei eine weitere Datenbank, die auf die Arbeiten von Tanaka, Euerby und Peterson basieren, genannt (ACD Labs Column Selection Database): http://www.acdlabs.com/products/adh/chrom/chromproc/index.php#colsel. Diese Datenbanken sind eine große Hilfe sich im Dschungel der HPLC-Säulen zurecht zu finden.

Abbildung: Bildschirmkopie der PQRI-Datenbank
Literatur:
[1] R. K. Iler, The Chemistry of Silica, Wiley, New York (1979)
[2] J.J. Kirkland, Performance of Zipax Controlled Surface Porosity Support in High-Speed Liquid Chromatography J Chromatogr Sci (1972) 10 (3): 129-137.
[3] C. Horvàth, S. R. Lipsky, J. Chromatogr. Sci., 7 (1969) 109 -116
[4] K.K. Unger, N. Tanaka, E. Machtjevas, Monolithic Silicas in Separation Science, Wiley-VCH (2011)
[5] P. G. Dietrich, J. Falkenhagen, R. Bertram, Verfahren zur Herstellung von Aluminiumoxid dotiertem Kiselgel zur chromatographischen Trennung freier Basen, deutsches Patent, DE4342493B4 (2006)
[6] S. Kromidas (Hsg), HPLC richtig optimiert, Wiley-VCH (2006)
[7] U.D. Neue, HPLC columns, Wiley-VCH (1997)
[8] S. Kromidas, Eigenschaften von kommerziellen HPLC-RP-Säulen im Vergleich, Pirrot Verlag (2002)
[9] H. Engelhardt, M. Jungheim, Chromatographia, 29 (1990) 59
[10] H. Engelhardt, M. Arangio, T. Lobert, LC/GC Int., 10(12) (1997) 803-812
[11] K. Kimata, K. Iwaguchi, S. Onishi, K. Jinno, R. Eksteen, K. Hosoya, M. Araki, N. Tanaka, J. Chromatogr. Sci., 27 (1989) 721- 728
[12] L. C. Sander, S. A. Wise, Anal. Chem., 56 (1984) 504-510
[13] L. C. Sander, S. A. Wise, Anal. Chem., 59 (1987) 2309-2313
[14] L. C. Sander, S. A. Wise, Anal. Chem., 67 (1995) 3284-3292
[15] U.D. Neue, J Sep Sci,30 (2007) 164ff
[16] P. G. Stevenson, K. J. Mayfield, A. Solivena, G. R. Dennisa, F. Gritti, G. Guiochon, R. A. Shalliker, Journal of Chromatography A, 1217 (2010) 5358–5364
[17] S. Nyiredy, Z. Szucs, L. Szepesy, Journal of Chromatography A, 1157 (2007), 122-130
[18] S. Nyiredy, Z. Szucs, L. Szepesy, Chromatographia 2009, 69, 609-614
[19] G. Socherl, R. Nussbaum, K. Rissler, E. Lankmayr, Chromatographia 54 (2001) 65-70
[20] K. Tanaka,M. Y. Ding, M. I. H. Helaleh, H. Taoda, H. Takahashi, W. Hu, K. Hasebe, P. R. Haddad, J. Fritz, C. Sarzanini, Journal of Chromatography A, 956 (2002) 209-214
[21] A.J. Alpert, Journal of Chromatography, 499 (1990) 177–196.
[22] S. M. Melnikov, A. Höltzel, A. Seidel-Morgenstern, U. Tallarek, Angew. Chem. 124 (2012) 6355 –6358
[23] J. Kriz, E. Adamcov, J. H. Knox, J. Hora, Journal of Chromatography, 663 (1995) 151ff
[24] L. R. Snyder, J. W. Dolan, P. W. Carr, J. Chromatogr. A, 1060 (2004) 77-116
[25] L. R. Snyder, J. W. Dolan, P. W. Car, Anal. Chem., 79 (2007) 3255-3261
[26] L. R. Snyder , A. Maule, A. Heebsch, R. Cuellar, S. Paulson, J. Carrano, L. Wrisley,, C. C. Chan, N. Pearson, J. W. Dolan and J. Gilroy, J. Chromatogr. A, 1057 (2004) 49-57
[27] J. W. Dolan, A. Maule, L. Wrisley,, C. C. Chan, M. Angod, C. Lunte, R. Krisko, J. Winston, B. Homeierand, D. M. McCalley and L. R. Snyder, J. Chromatogr. A, 1057 (2004) 59-74